

Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment
- PMID: 32810438
- PMCID: PMC7405822
- DOI: 10.1016/j.cell.2020.08.001
Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment
Abstract
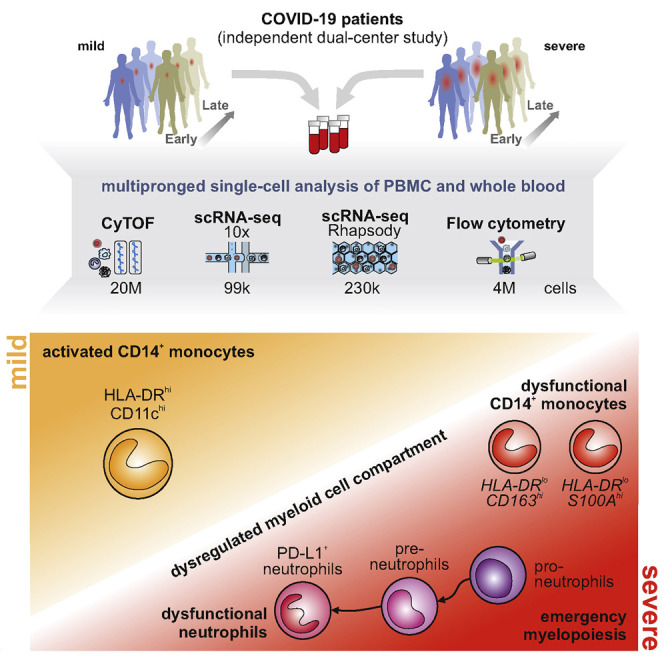
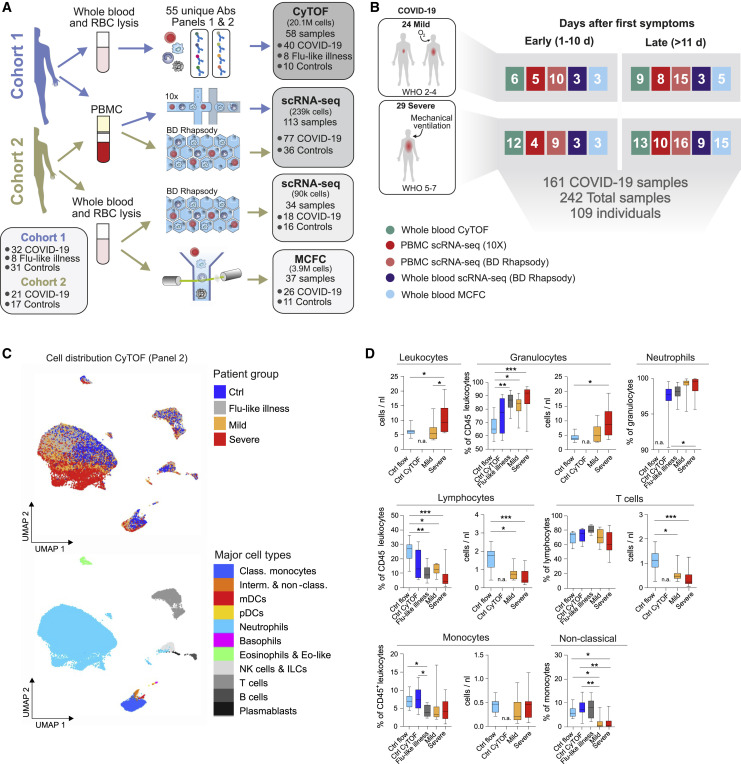
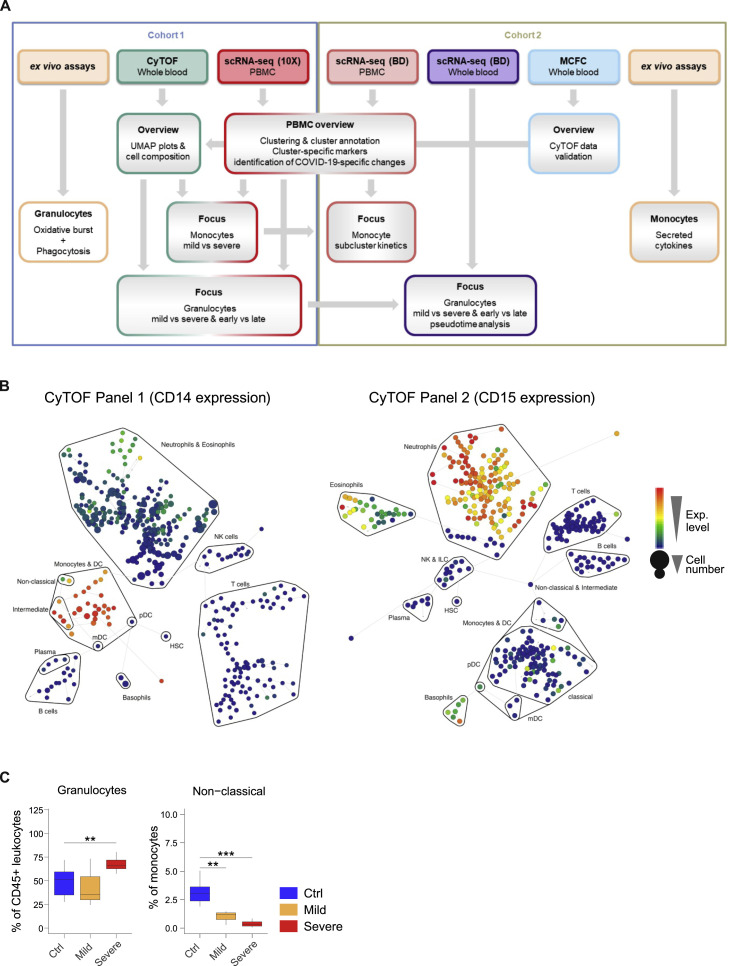
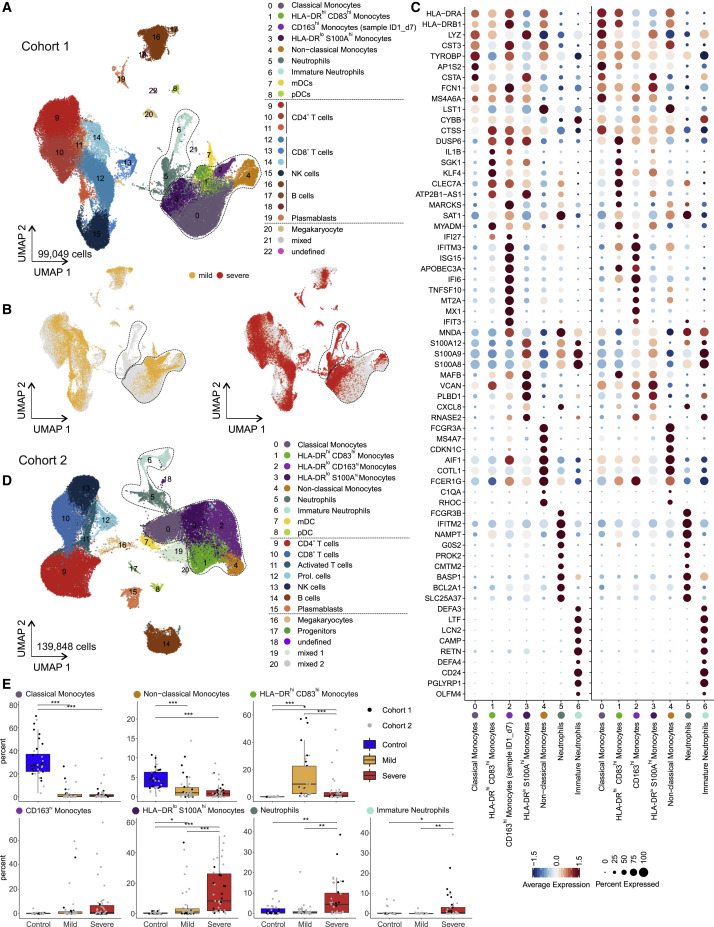
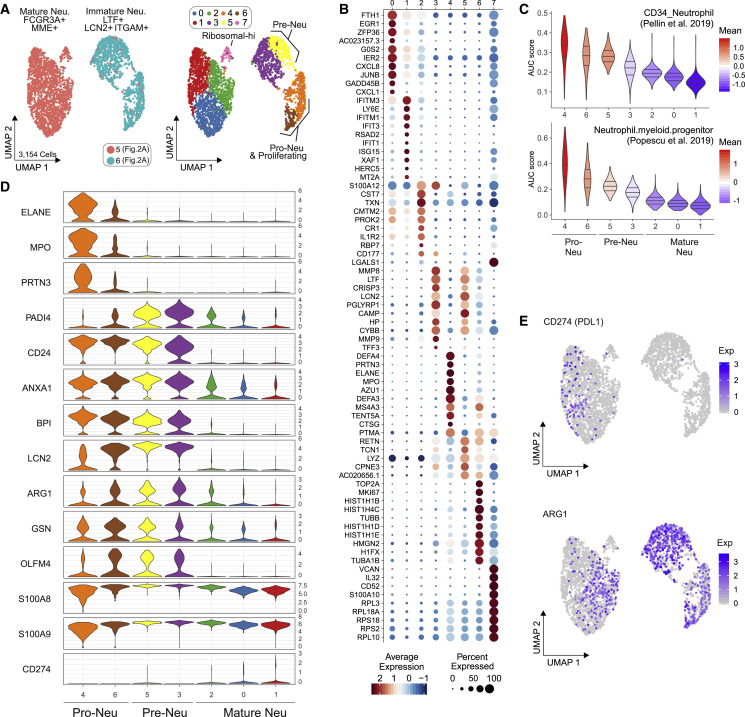
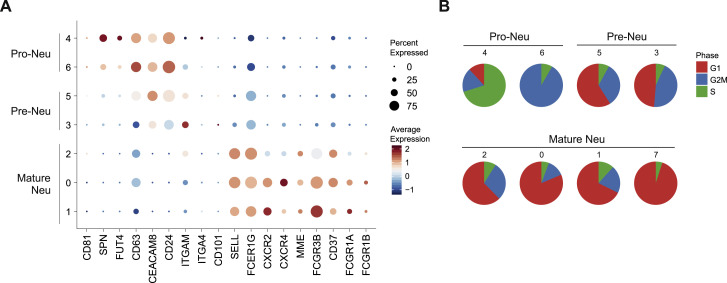
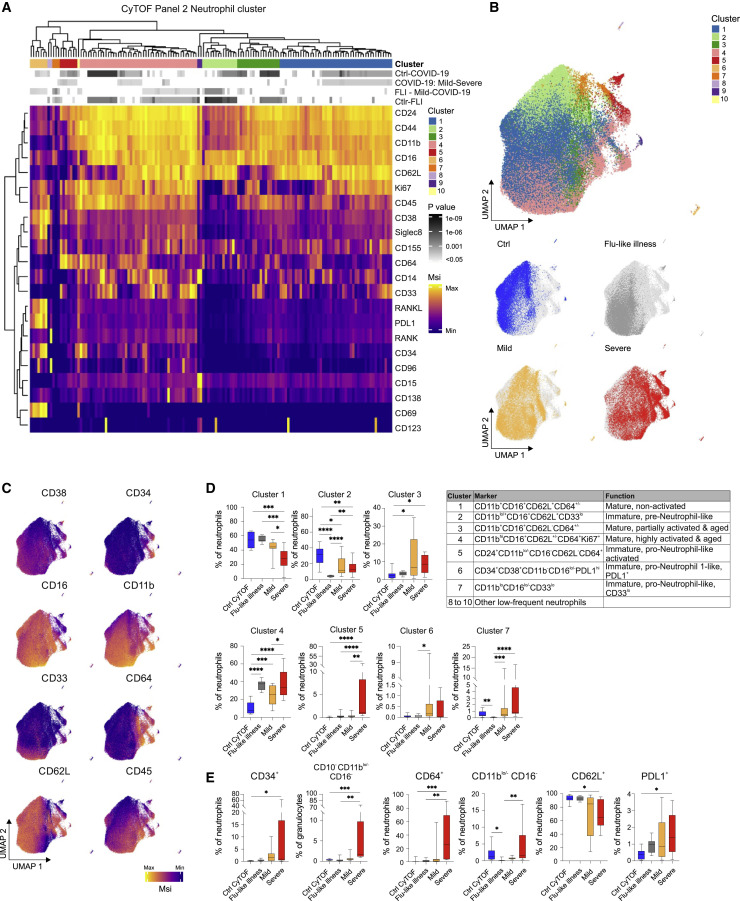
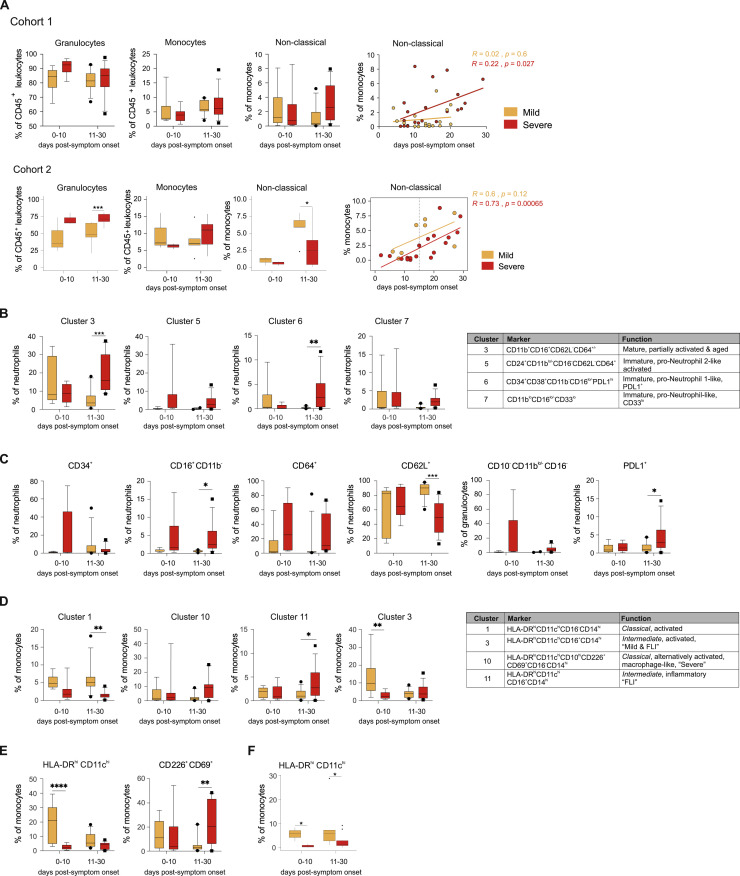
Coronavirus disease 2019 (COVID-19) is a mild to moderate respiratory tract infection, however, a subset of patients progress to severe disease and respiratory failure. The mechanism of protective immunity in mild forms and the pathogenesis of severe COVID-19 associated with increased neutrophil counts and dysregulated immune responses remain unclear. In a dual-center, two-cohort study, we combined single-cell RNA-sequencing and single-cell proteomics of whole-blood and peripheral-blood mononuclear cells to determine changes in immune cell composition and activation in mild versus severe COVID-19 (242 samples from 109 individuals) over time. HLA-DRhiCD11chi inflammatory monocytes with an interferon-stimulated gene signature were elevated in mild COVID-19. Severe COVID-19 was marked by occurrence of neutrophil precursors, as evidence of emergency myelopoiesis, dysfunctional mature neutrophils, and HLA-DRlo monocytes. Our study provides detailed insights into the systemic immune response to SARS-CoV-2 infection and reveals profound alterations in the myeloid cell compartment associated with severe COVID-19.
Keywords: COVID-19; SARS-CoV-2; dysfunctional neutrophils; emergency myelopoiesis; immune profiling; mass cytometry; monocytes; neutrophils; scRNA-seq.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
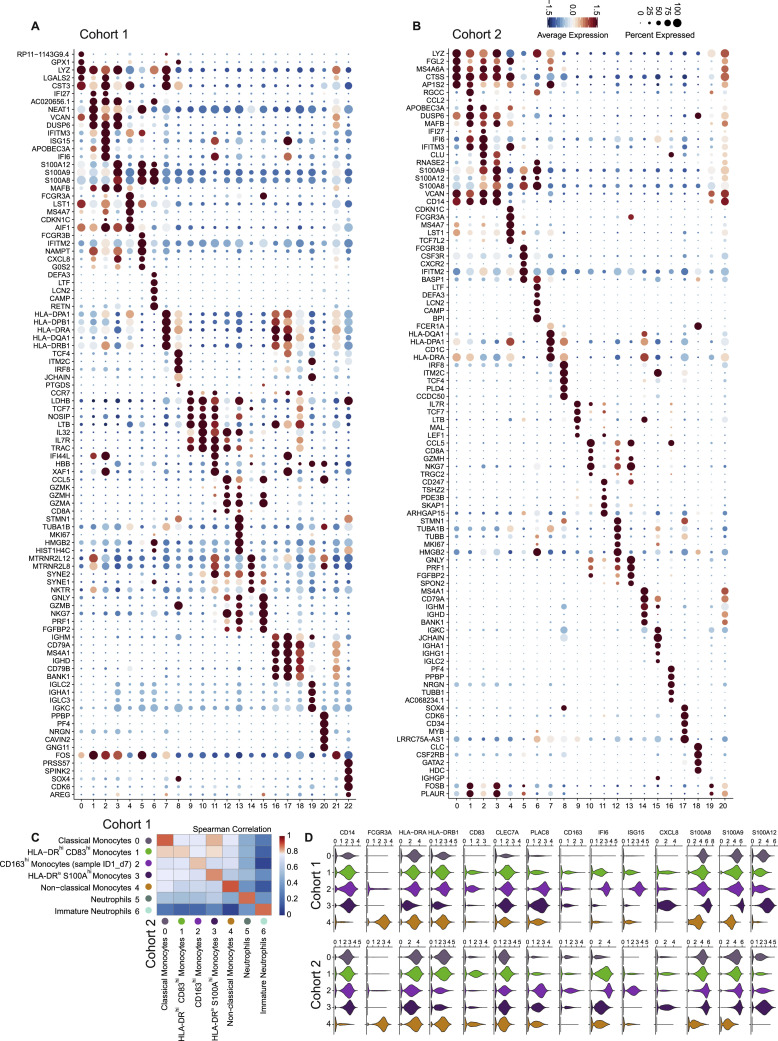
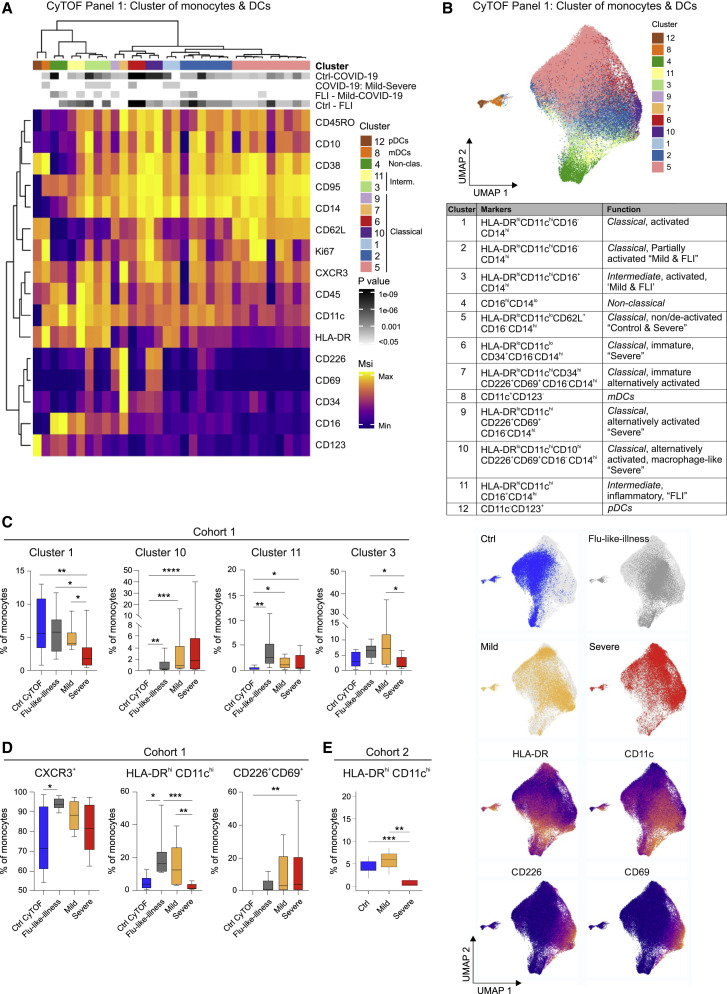
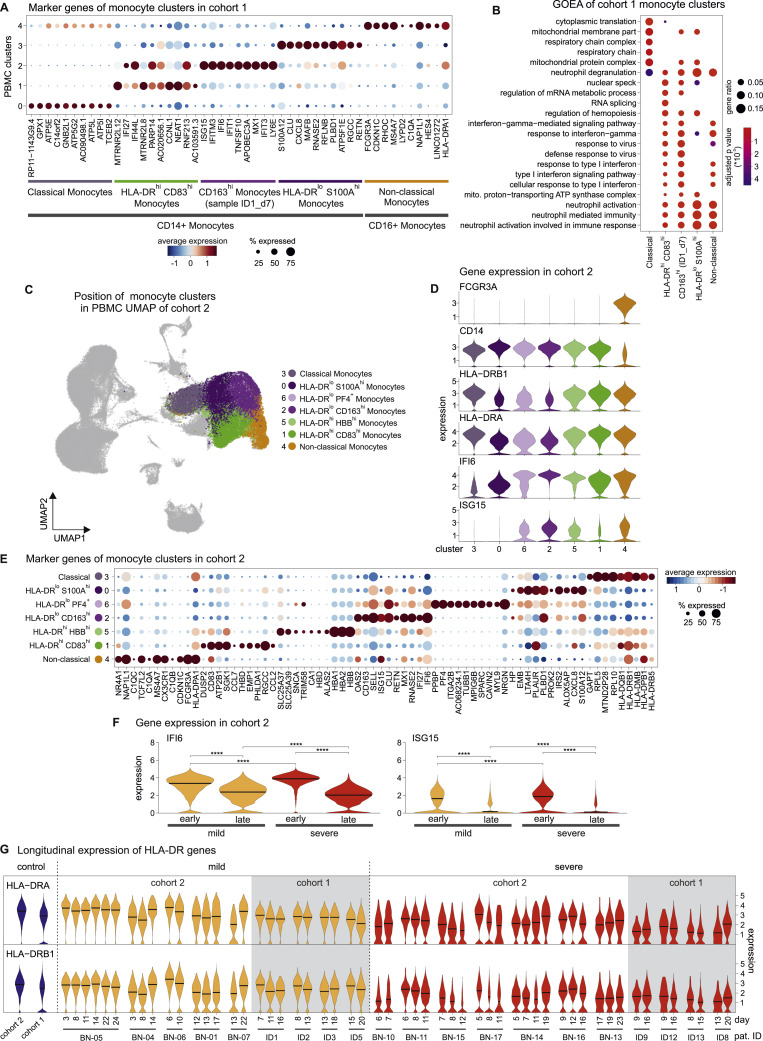
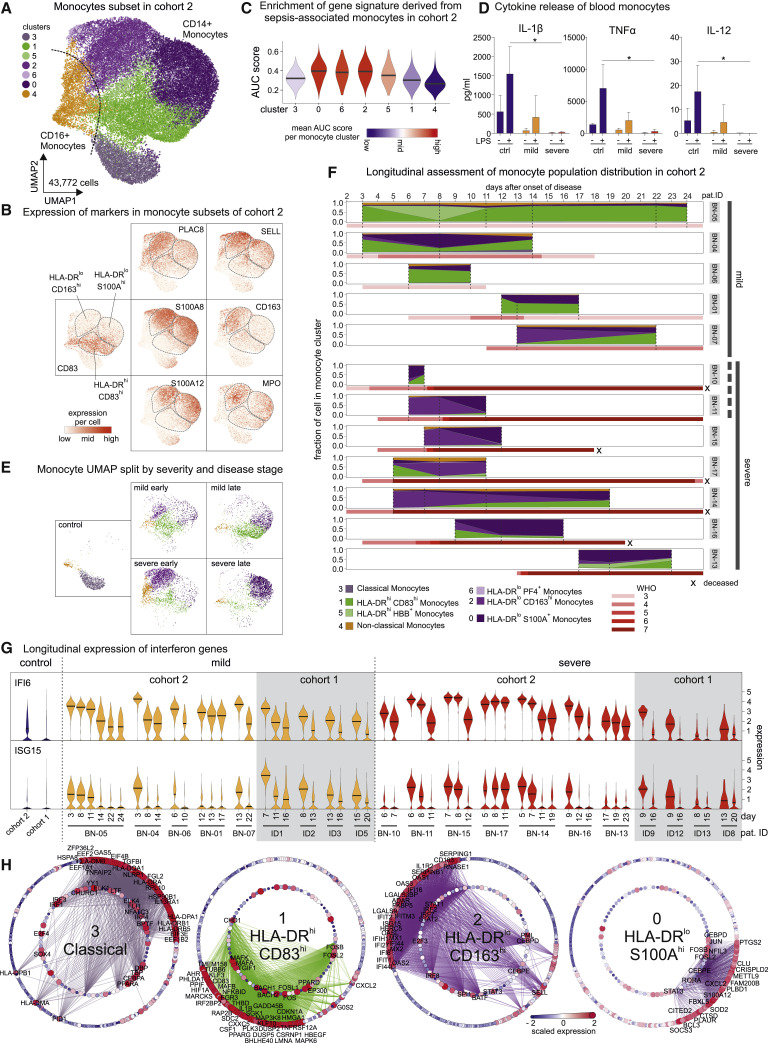
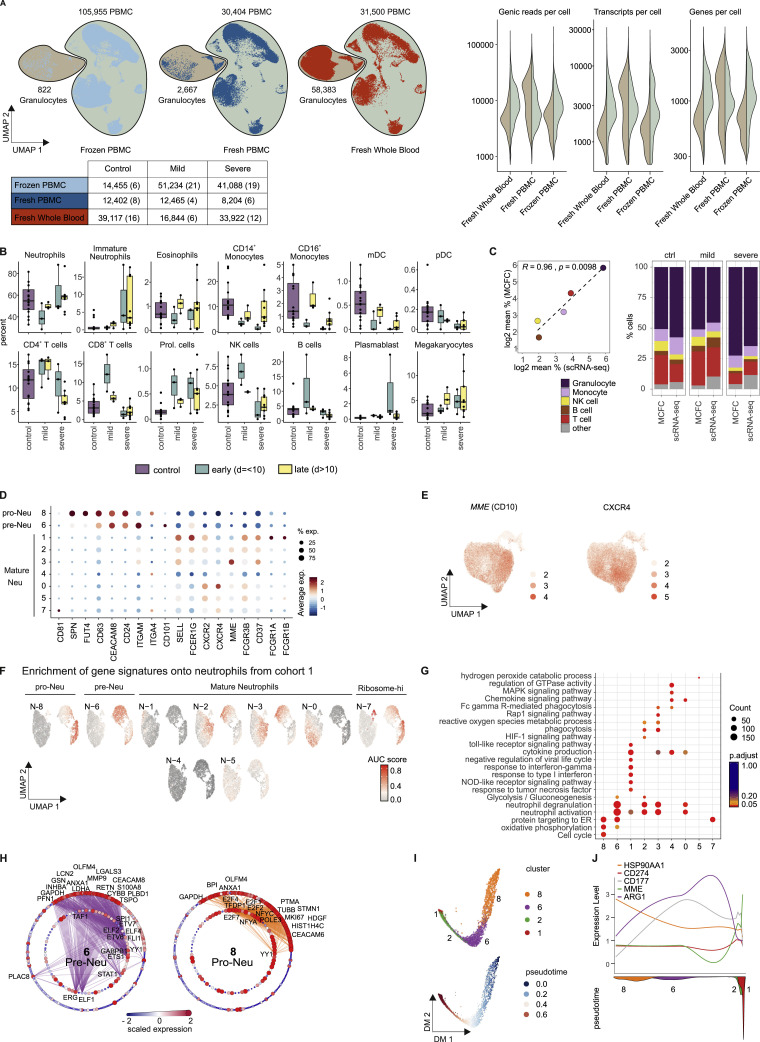
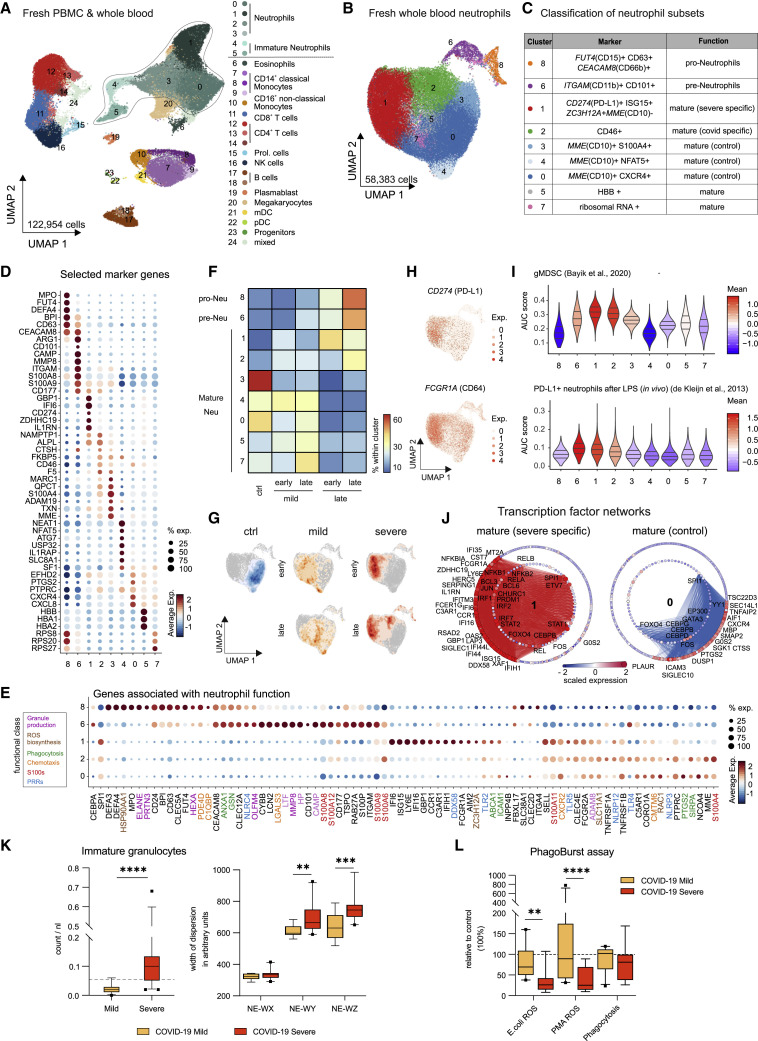
References
-
- Angerer P., Haghverdi L., Büttner M., Theis F.J., Marr C., Buettner F. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–1243. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
