

Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity
- PMID: 32814791
- PMCID: PMC7438523
- DOI: 10.1038/s41598-020-70812-6
Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity
Erratum in
-
Author Correction: Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity.Sci Rep. 2021 Oct 12;11(1):20568. doi: 10.1038/s41598-021-00133-9. Sci Rep. 2021. PMID: 34642347 Free PMC article. No abstract available.
Abstract
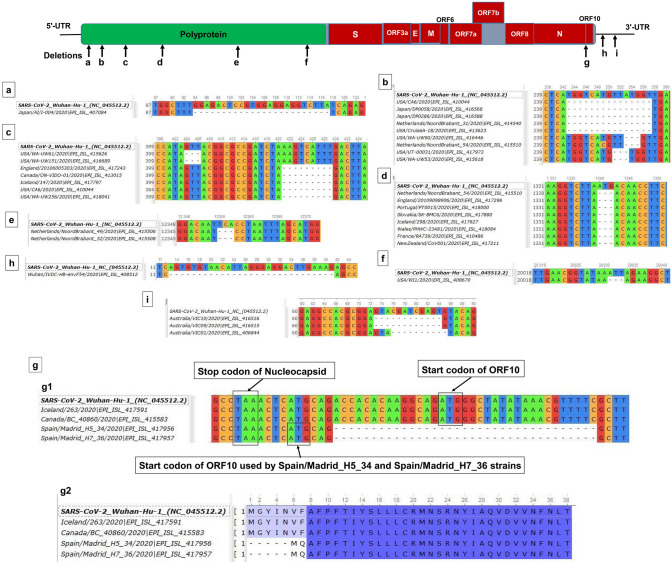
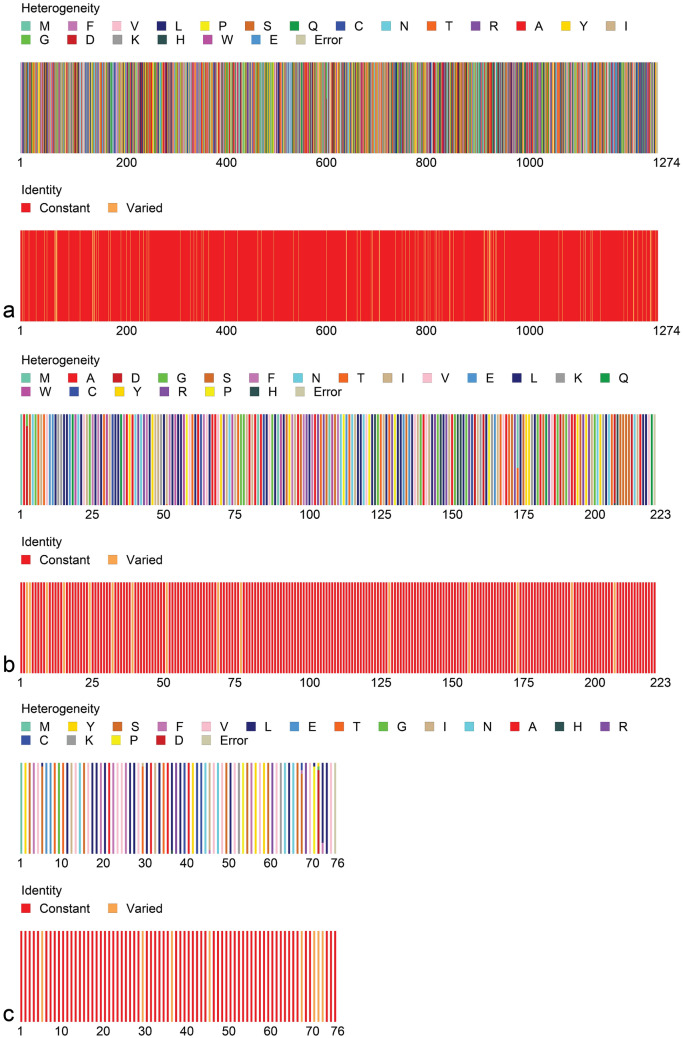
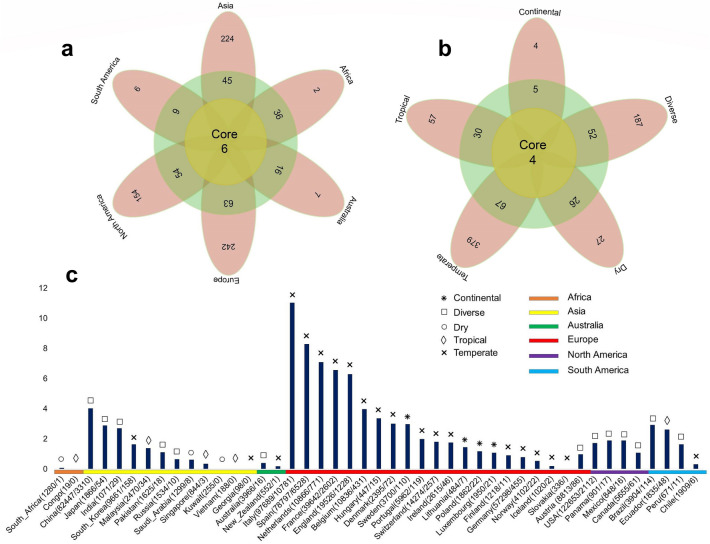
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), a novel evolutionary divergent RNA virus, is responsible for the present devastating COVID-19 pandemic. To explore the genomic signatures, we comprehensively analyzed 2,492 complete and/or near-complete genome sequences of SARS-CoV-2 strains reported from across the globe to the GISAID database up to 30 March 2020. Genome-wide annotations revealed 1,516 nucleotide-level variations at different positions throughout the entire genome of SARS-CoV-2. Moreover, nucleotide (nt) deletion analysis found twelve deletion sites throughout the genome other than previously reported deletions at coding sequence of the ORF8 (open reading frame), spike, and ORF7a proteins, specifically in polyprotein ORF1ab (n = 9), ORF10 (n = 1), and 3´-UTR (n = 2). Evidence from the systematic gene-level mutational and protein profile analyses revealed a large number of amino acid (aa) substitutions (n = 744), demonstrating the viral proteins heterogeneous. Notably, residues of receptor-binding domain (RBD) showing crucial interactions with angiotensin-converting enzyme 2 (ACE2) and cross-reacting neutralizing antibody were found to be conserved among the analyzed virus strains, except for replacement of lysine with arginine at 378th position of the cryptic epitope of a Shanghai isolate, hCoV-19/Shanghai/SH0007/2020 (EPI_ISL_416320). Furthermore, our results of the preliminary epidemiological data on SARS-CoV-2 infections revealed that frequency of aa mutations were relatively higher in the SARS-CoV-2 genome sequences of Europe (43.07%) followed by Asia (38.09%), and North America (29.64%) while case fatality rates remained higher in the European temperate countries, such as Italy, Spain, Netherlands, France, England and Belgium. Thus, the present method of genome annotation employed at this early pandemic stage could be a promising tool for monitoring and tracking the continuously evolving pandemic situation, the associated genetic variants, and their implications for the development of effective control and prophylaxis strategies.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
