

Epigenetic modification and therapeutic targets of diabetes mellitus
- PMID: 32815547
- PMCID: PMC7494983
- DOI: 10.1042/BSR20202160
Epigenetic modification and therapeutic targets of diabetes mellitus
Abstract
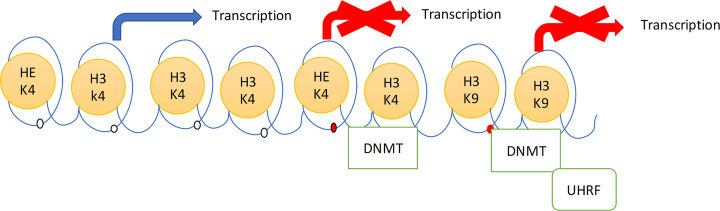
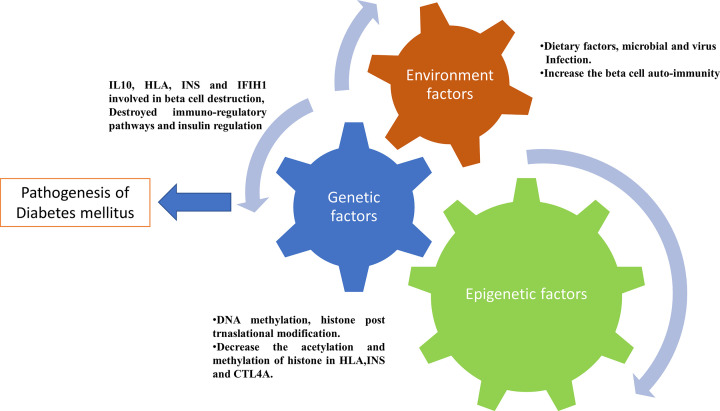
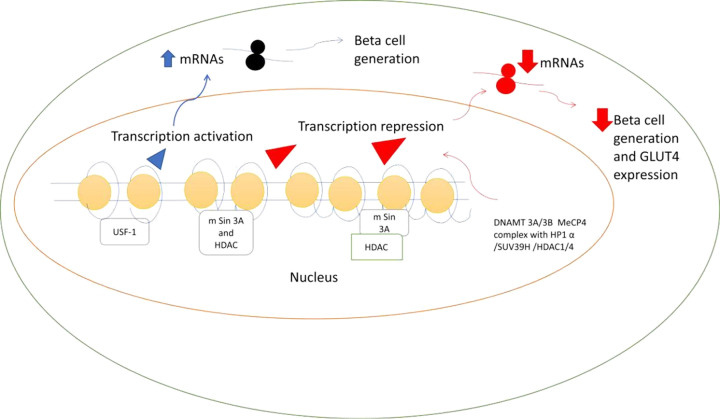
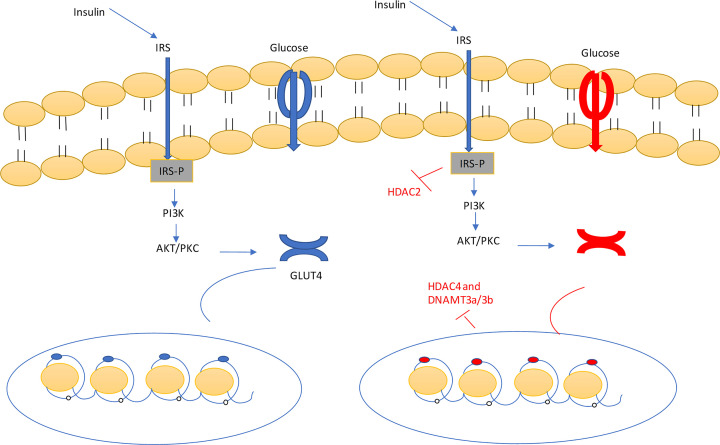
The prevalence of diabetes and its related complications are increasing significantly globally. Collected evidence suggested that several genetic and environmental factors contribute to diabetes mellitus. Associated complications such as retinopathy, neuropathy, nephropathy and other cardiovascular complications are a direct result of diabetes. Epigenetic factors include deoxyribonucleic acid (DNA) methylation and histone post-translational modifications. These factors are directly related with pathological factors such as oxidative stress, generation of inflammatory mediators and hyperglycemia. These result in altered gene expression and targets cells in the pathology of diabetes mellitus without specific changes in a DNA sequence. Environmental factors and malnutrition are equally responsible for epigenetic states. Accumulated evidence suggested that environmental stimuli alter the gene expression that result in epigenetic changes in chromatin. Recent studies proposed that epigenetics may include the occurrence of 'metabolic memory' found in animal studies. Further study into epigenetic mechanism might give us new vision into the pathogenesis of diabetes mellitus and related complication thus leading to the discovery of new therapeutic targets. In this review, we discuss the possible epigenetic changes and mechanism that happen in diabetes mellitus type 1 and type 2 separately. We highlight the important epigenetic and non-epigenetic therapeutic targets involved in the management of diabetes and associated complications.
Keywords: Molecular Targets; diabetes mellitus; epigenetics.
© 2020 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
Similar articles
-
Epigenetic Regulation of β Cell Identity and Dysfunction.Front Endocrinol (Lausanne). 2021 Sep 24;12:725131. doi: 10.3389/fendo.2021.725131. eCollection 2021. Front Endocrinol (Lausanne). 2021. PMID: 34630329 Free PMC article. Review.
-
Epigenetic mechanisms in diabetic complications and metabolic memory.Diabetologia. 2015 Mar;58(3):443-55. doi: 10.1007/s00125-014-3462-y. Epub 2014 Dec 7. Diabetologia. 2015. PMID: 25481708 Free PMC article. Review.
-
Epigenetic Regulation Associated With Sirtuin 1 in Complications of Diabetes Mellitus.Front Endocrinol (Lausanne). 2021 Jan 18;11:598012. doi: 10.3389/fendo.2020.598012. eCollection 2020. Front Endocrinol (Lausanne). 2021. PMID: 33537003 Free PMC article. Review.
-
Epigenomics in stress tolerance of plants under the climate change.Mol Biol Rep. 2023 Jul;50(7):6201-6216. doi: 10.1007/s11033-023-08539-6. Epub 2023 Jun 9. Mol Biol Rep. 2023. PMID: 37294468 Review.
-
Epigenetic modifications: An important mechanism in diabetic disturbances.Postepy Hig Med Dosw (Online). 2017 Nov 29;71(0):960-974. doi: 10.5604/01.3001.0010.6156. Postepy Hig Med Dosw (Online). 2017. PMID: 29225202 Review.
Cited by
-
miRNA and leptin signaling in metabolic diseases and at extreme environments.Pharmacol Res Perspect. 2024 Aug;12(4):e1248. doi: 10.1002/prp2.1248. Pharmacol Res Perspect. 2024. PMID: 39017237 Free PMC article. Review.
-
Epigenetic basis of diabetic vasculopathy.Front Endocrinol (Lausanne). 2022 Dec 9;13:989844. doi: 10.3389/fendo.2022.989844. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36568089 Free PMC article. Review.
-
Histone Modifications and Non-Coding RNAs: Mutual Epigenetic Regulation and Role in Pathogenesis.Int J Mol Sci. 2022 May 22;23(10):5801. doi: 10.3390/ijms23105801. Int J Mol Sci. 2022. PMID: 35628612 Free PMC article. Review.
-
7-Ketocholesterol accelerates pancreatic β-cell senescence by inhibiting the SIRT1/CDK4-Rb-E2F1 signaling pathway.Islets. 2023 Dec 31;15(1):2219105. doi: 10.1080/19382014.2023.2219105. Islets. 2023. PMID: 37265106 Free PMC article.
-
The potential protective effect of aqueous extract of Acanthophyllum glandulosum root on Streptozotocin-induced diabetes in mice.J Diabetes Metab Disord. 2023 Jul 12;22(2):1231-1243. doi: 10.1007/s40200-023-01238-w. eCollection 2023 Dec. J Diabetes Metab Disord. 2023. PMID: 37975083 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical