

BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition
- PMID: 32816843
- PMCID: PMC7892644
- DOI: 10.1158/2159-8290.CD-20-0142
BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition
Abstract
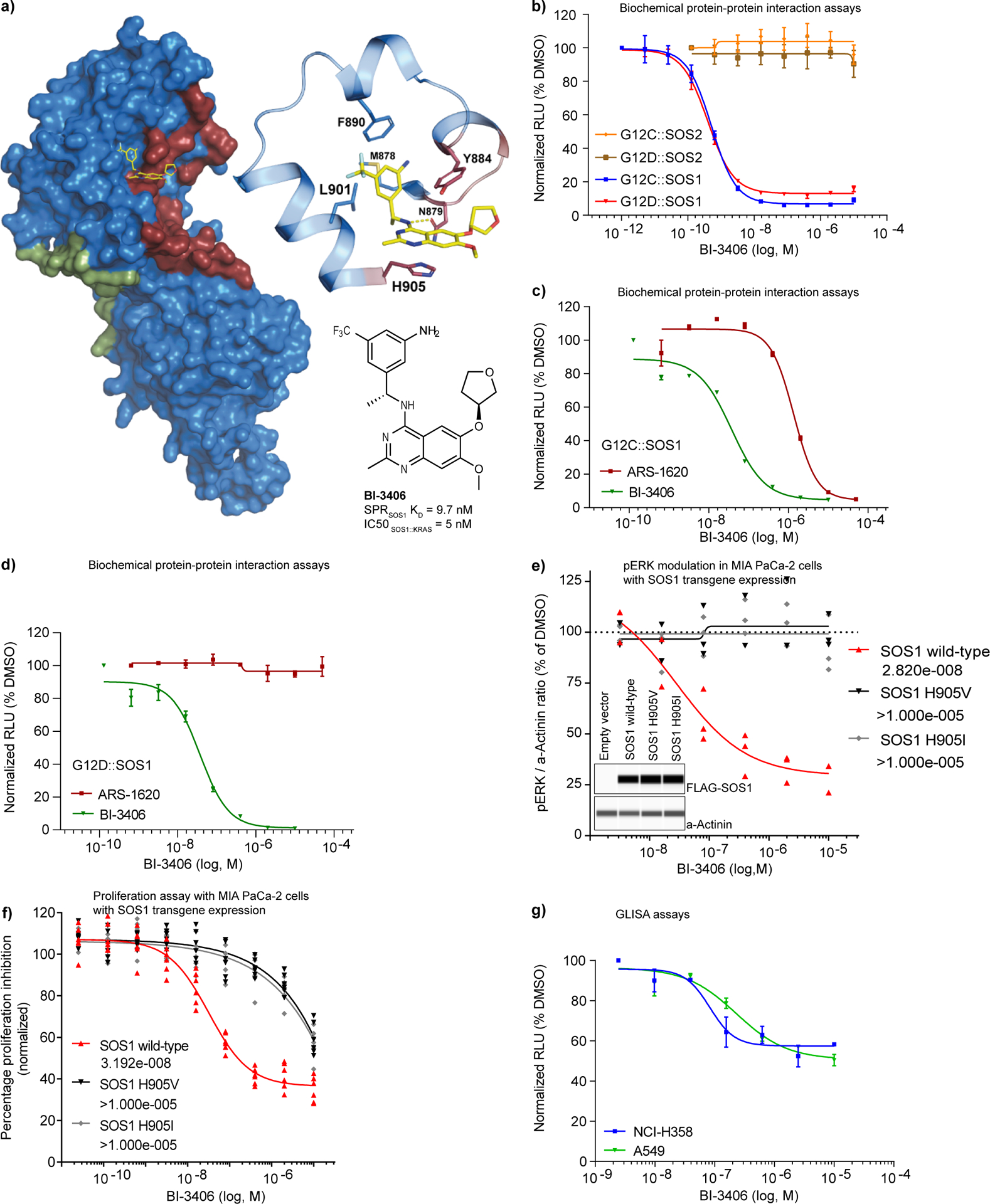
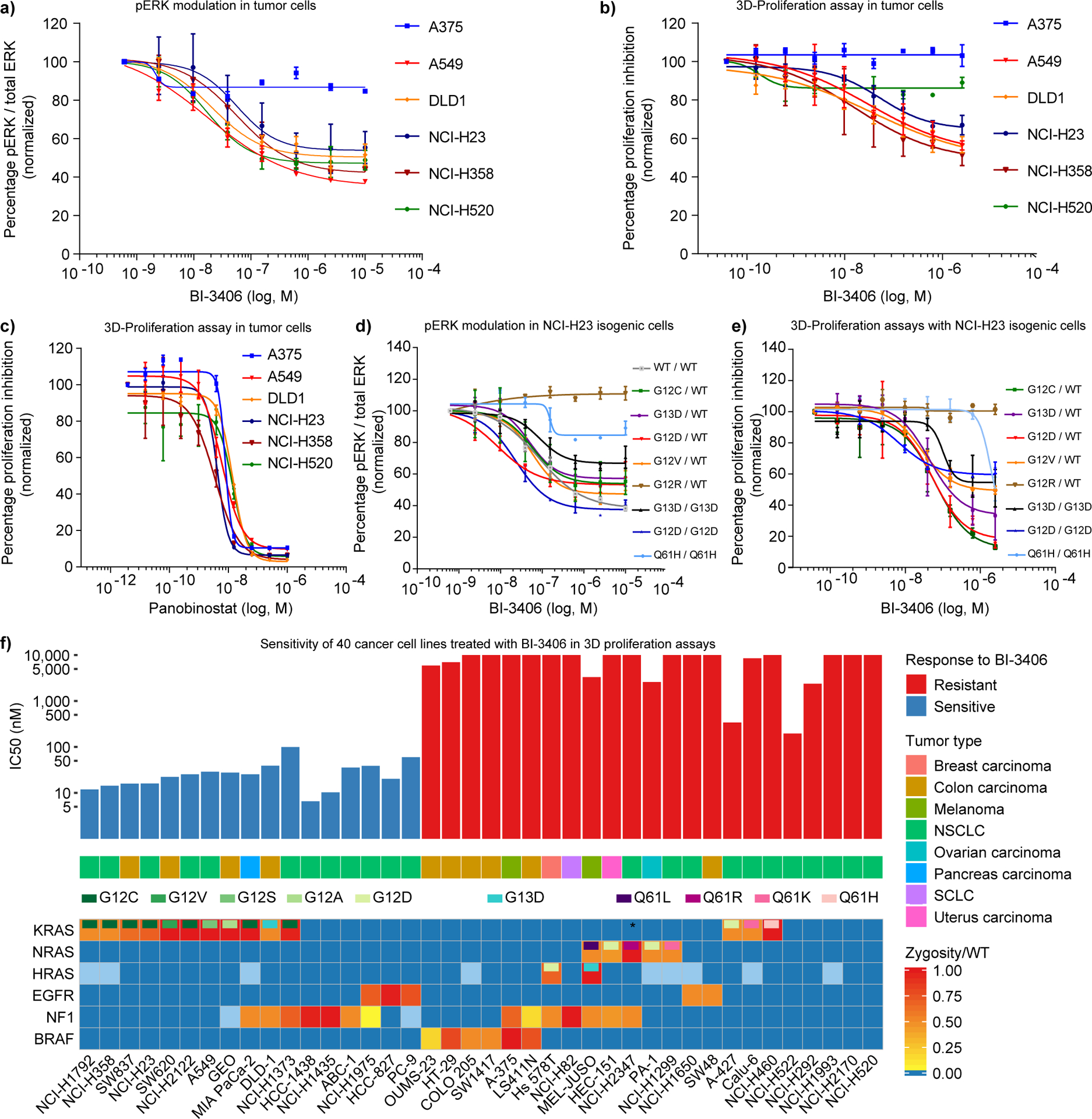
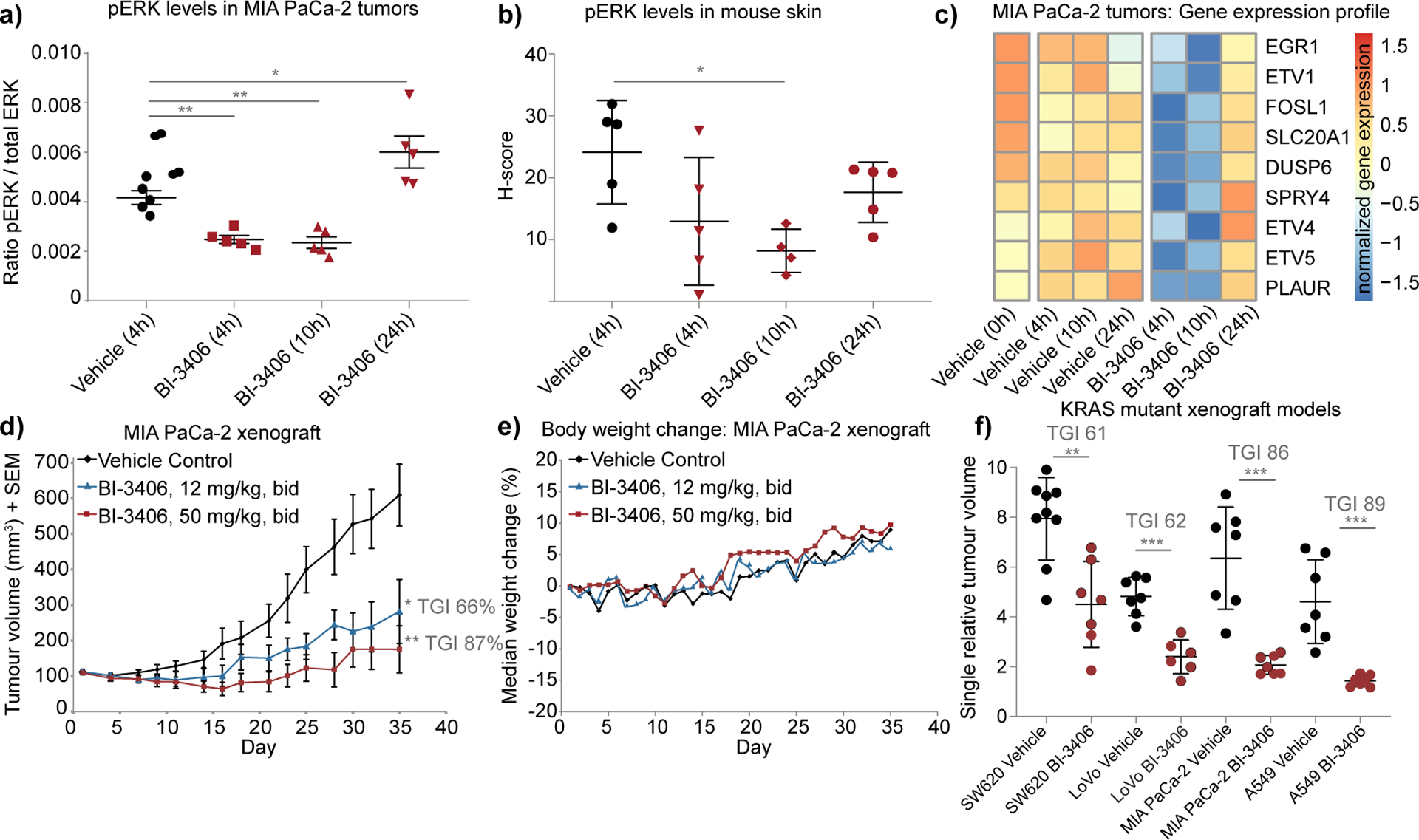
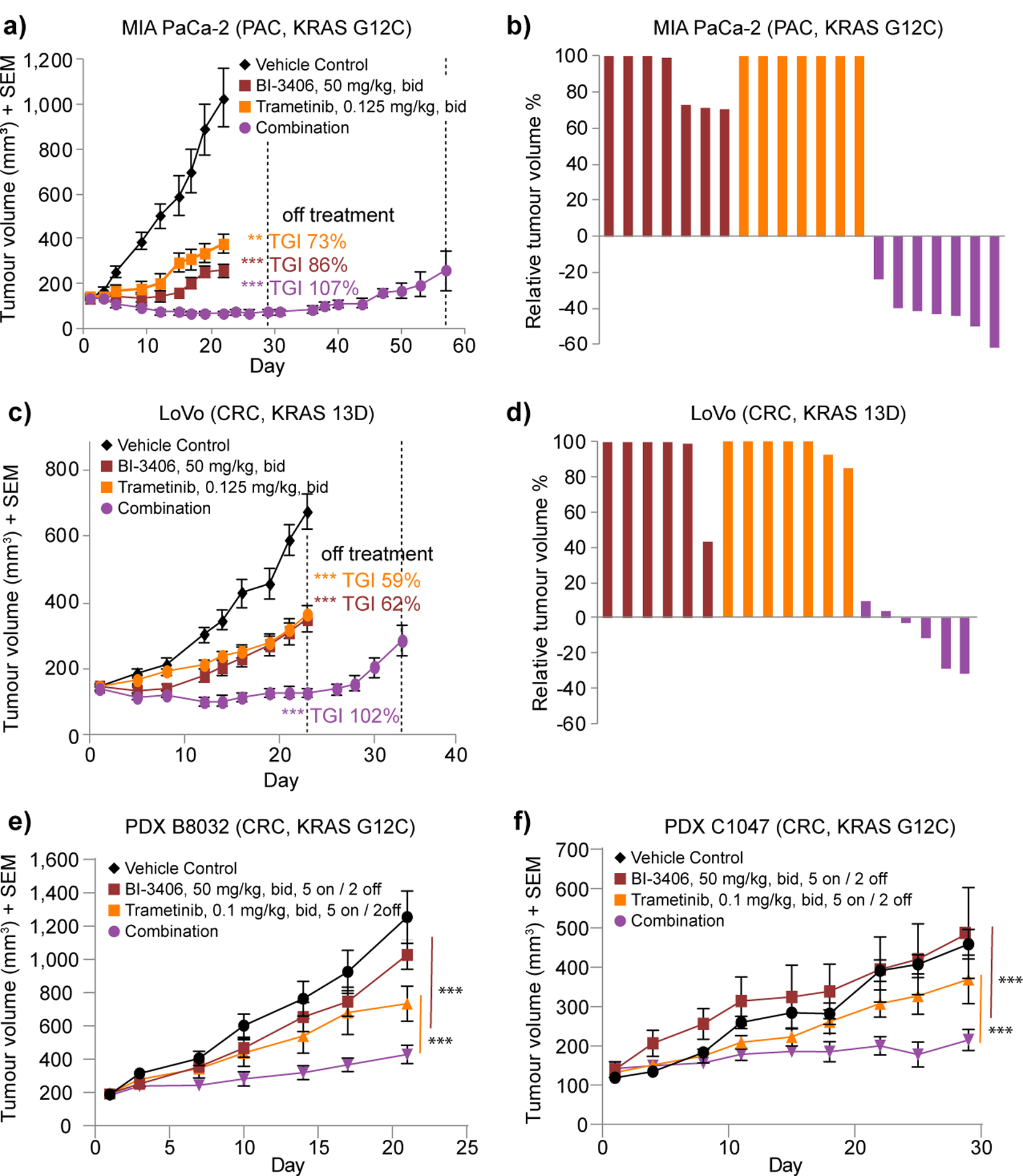
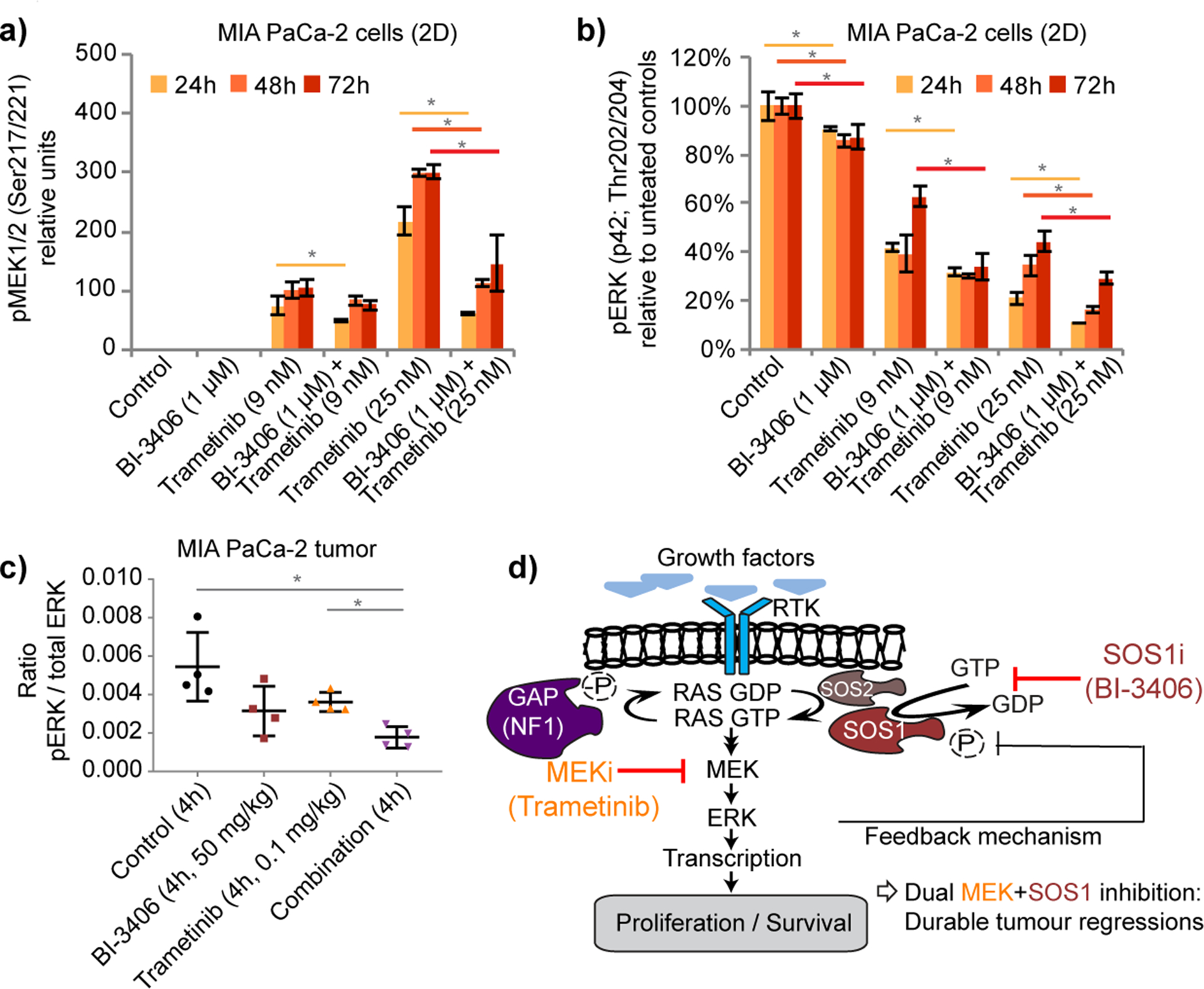
KRAS is the most frequently mutated driver of pancreatic, colorectal, and non-small cell lung cancers. Direct KRAS blockade has proved challenging, and inhibition of a key downstream effector pathway, the RAF-MEK-ERK cascade, has shown limited success because of activation of feedback networks that keep the pathway in check. We hypothesized that inhibiting SOS1, a KRAS activator and important feedback node, represents an effective approach to treat KRAS-driven cancers. We report the discovery of a highly potent, selective, and orally bioavailable small-molecule SOS1 inhibitor, BI-3406, that binds to the catalytic domain of SOS1, thereby preventing the interaction with KRAS. BI-3406 reduces formation of GTP-loaded RAS and limits cellular proliferation of a broad range of KRAS-driven cancers. Importantly, BI-3406 attenuates feedback reactivation induced by MEK inhibitors and thereby enhances sensitivity of KRAS-dependent cancers to MEK inhibition. Combined SOS1 and MEK inhibition represents a novel and effective therapeutic concept to address KRAS-driven tumors. SIGNIFICANCE: To date, there are no effective targeted pan-KRAS therapies. In-depth characterization of BI-3406 activity and identification of MEK inhibitors as effective combination partners provide an attractive therapeutic concept for the majority of KRAS-mutant cancers, including those fueled by the most prevalent mutant KRAS oncoproteins, G12D, G12V, G12C, and G13D.See related commentary by Zhao et al., p. 17.This article is highlighted in the In This Issue feature, p. 1.
©2020 American Association for Cancer Research.
Conflict of interest statement
Figures
Comment in
-
Suppressing Nucleotide Exchange to Inhibit KRAS-Mutant Tumors.Cancer Discov. 2021 Jan;11(1):17-19. doi: 10.1158/2159-8290.CD-20-1331. Cancer Discov. 2021. PMID: 34003780
Comment on
-
Suppressing Nucleotide Exchange to Inhibit KRAS-Mutant Tumors.Cancer Discov. 2021 Jan;11(1):17-19. doi: 10.1158/2159-8290.CD-20-1331. Cancer Discov. 2021. PMID: 34003780
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
