

Molecular Characterization of Influenza C Viruses from Outbreaks in Hong Kong SAR, China
- PMID: 32817211
- PMCID: PMC7565627
- DOI: 10.1128/JVI.01051-20
Molecular Characterization of Influenza C Viruses from Outbreaks in Hong Kong SAR, China
Abstract
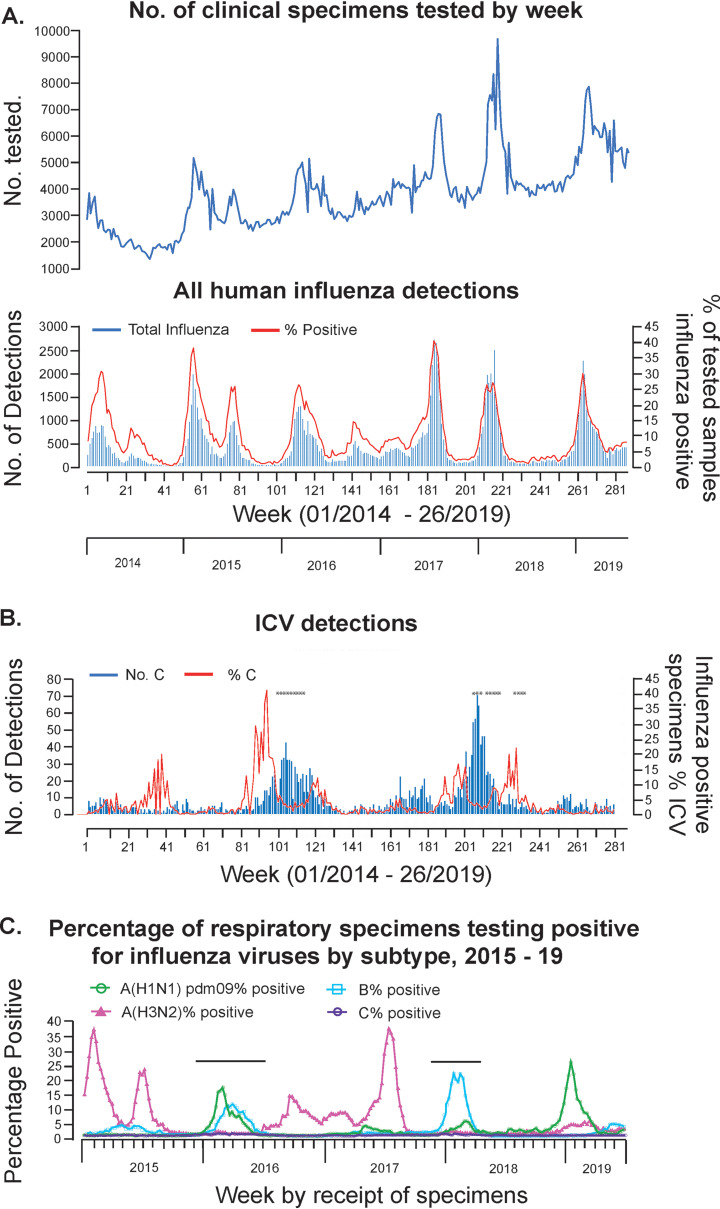
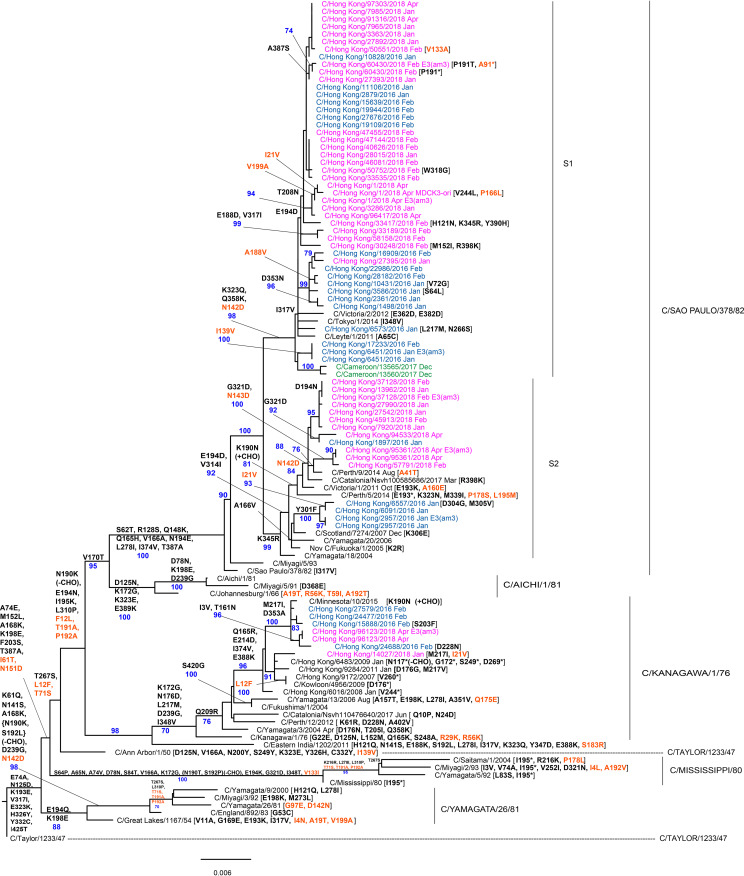
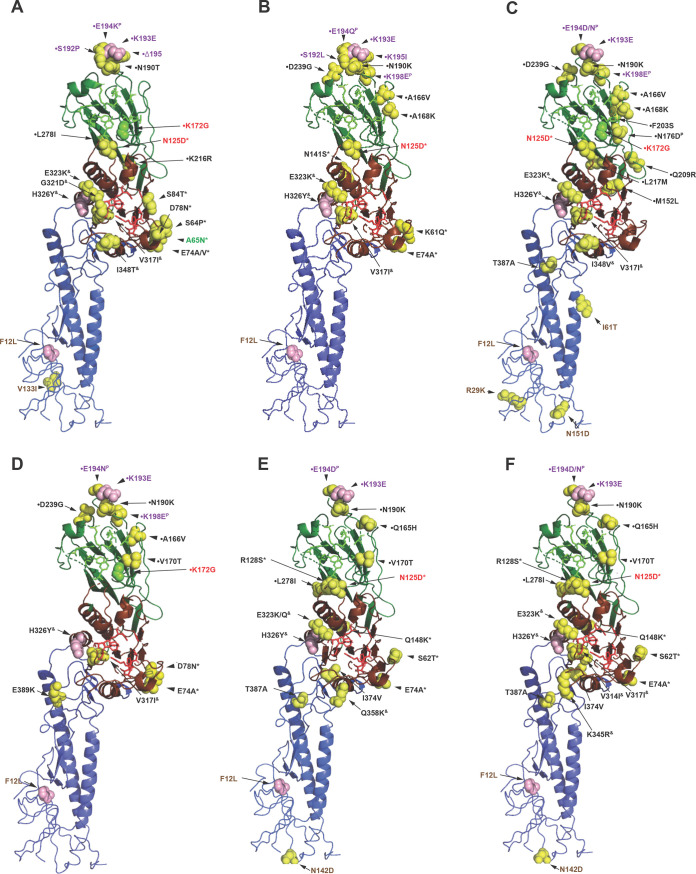
In 2014, the Centre for Health Protection in Hong Kong introduced screening for influenza C virus (ICV) as part of its routine surveillance for infectious agents in specimens collected from patients presenting with symptoms of respiratory viral infection, including influenza-like illness (ILI). A retrospective analysis of ICV detections up to week 26 of 2019 revealed persistent low-level circulation, with two outbreaks having occurred in the winters of 2015 to 2016 and 2017 to 2018. These outbreaks occurred at the same time as, and were dwarfed by, seasonal epidemics of influenza types A and B. Gene sequencing studies on stored ICV-positive clinical specimens from the two outbreaks have shown that the hemagglutinin-esterase (HE) genes of the viruses fall into two of the six recognized genetic lineages (represented by C/Kanagawa/1/76 and C/São Paulo/378/82), with there being significant genetic drift compared to earlier circulating viruses within both lineages. The location of a number of encoded amino acid substitutions in hemagglutinin-esterase fusion (HEF) glycoproteins suggests that antigenic drift may also have occurred. Observations of ICV outbreaks in other countries, with some of the infections being associated with severe disease, indicates that ICV infection has the potential to have significant clinical and health care impacts in humans.IMPORTANCE Influenza C virus infection of humans is common, and reinfection can occur throughout life. While symptoms are generally mild, severe disease cases have been reported, but knowledge of the virus is limited, as little systematic surveillance for influenza C virus is conducted and the virus cannot be studied by classical virologic methods because it cannot be readily isolated in laboratories. A combination of systematic surveillance in Hong Kong SAR, China, and new gene sequencing methods has been used in this study to assess influenza C virus evolution and provides evidence for a 2-year cycle of disease outbreaks. The results of studies like that reported here are key to developing an understanding of the impact of influenza C virus infection in humans and how virus evolution might be associated with epidemics.
Keywords: genome sequencing; influenza C virus; outbreak surveillance; virus evolution.
Copyright © 2020 Daniels et al.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
