

No evidence for distinct types in the evolution of SARS-CoV-2
- PMID: 32817804
- PMCID: PMC7197565
- DOI: 10.1093/ve/veaa034
No evidence for distinct types in the evolution of SARS-CoV-2
Abstract
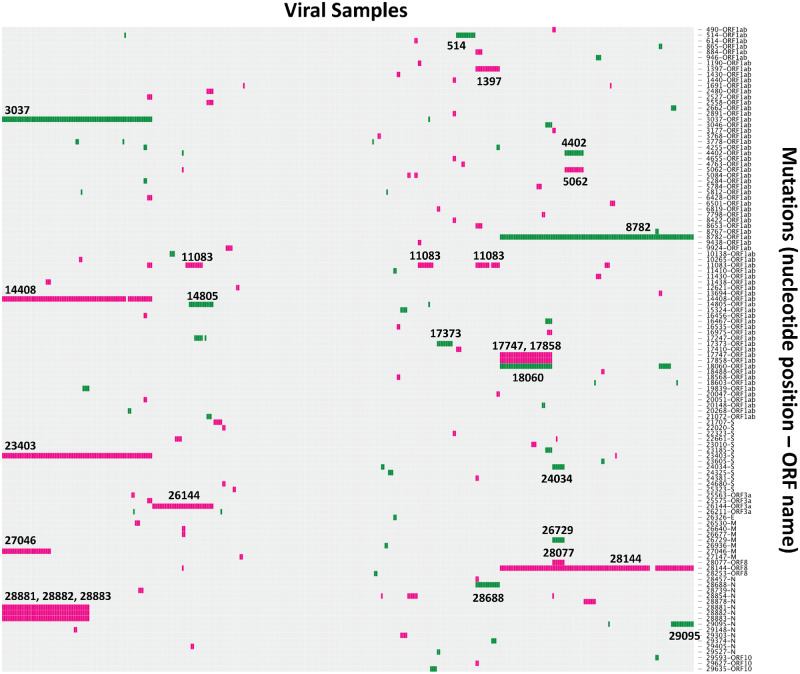
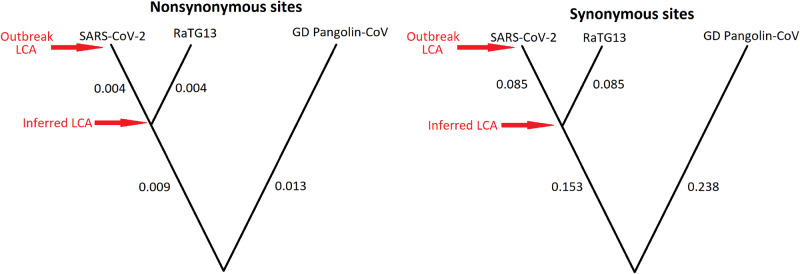
A recent study by Tang et al. claimed that two major types of severe acute respiratory syndrome-coronavirus-2 (CoV-2) had evolved in the ongoing CoV disease-2019 pandemic and that one of these types was more 'aggressive' than the other. Given the repercussions of these claims and the intense media coverage of these types of articles, we have examined in detail the data presented by Tang et al., and show that the major conclusions of that paper cannot be substantiated. Using examples from other viral outbreaks, we discuss the difficulty in demonstrating the existence or nature of a functional effect of a viral mutation, and we advise against overinterpretation of genomic data during the pandemic.
Keywords: COVID-19; SARS-CoV-2; adaptation.
© The Author(s) 2020. Published by Oxford University Press.
Figures

References
-
- Bhattacharya T. et al. (2007) ‘Founder Effects in the Assessment of HIV Polymorphisms and HLA Allele Associations’, Science, 315: 1583–6. - PubMed
-
- Boni M. F. et al. (2020) ‘Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic’, bioRxiv. doi:10.1101/2020.03.30.015008. - PubMed
-
- Deng X. et al. (2020) ‘A Genomic Survey of SARS-CoV-2 Reveals Multiple Introductions into Northern California without a Predominant Lineage’, medRxiv. doi:10.1101/2020.03.27.20044925.
LinkOut - more resources
Full Text Sources
Miscellaneous
