

This is a preprint.
Characterizing SARS-CoV-2 mutations in the United States
- PMID: 32818213
- PMCID: PMC7430589
- DOI: 10.21203/rs.3.rs-49671/v1
Characterizing SARS-CoV-2 mutations in the United States
Update in
-
Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants.Commun Biol. 2021 Feb 15;4(1):228. doi: 10.1038/s42003-021-01754-6. Commun Biol. 2021. PMID: 33589648 Free PMC article.
Abstract
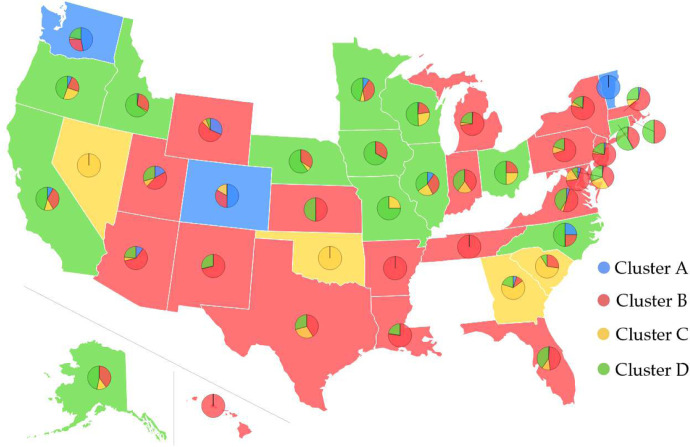
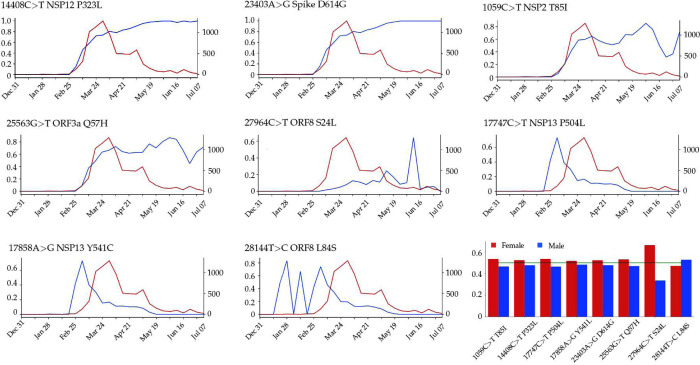
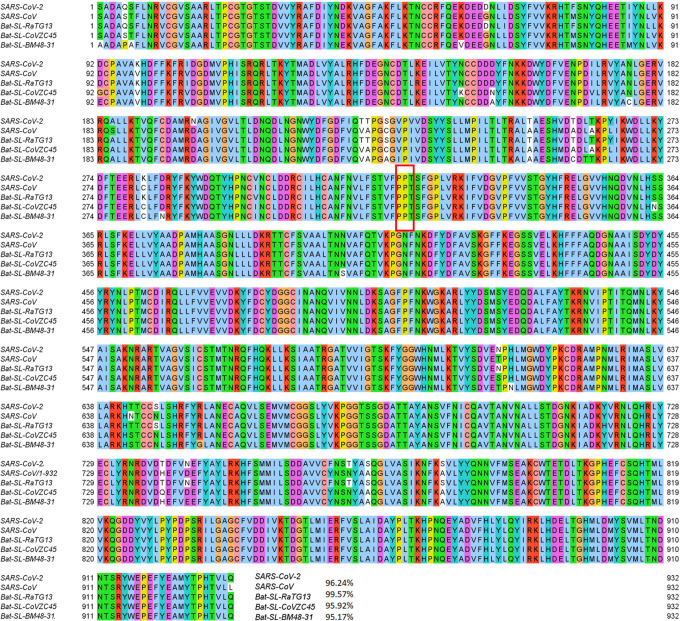
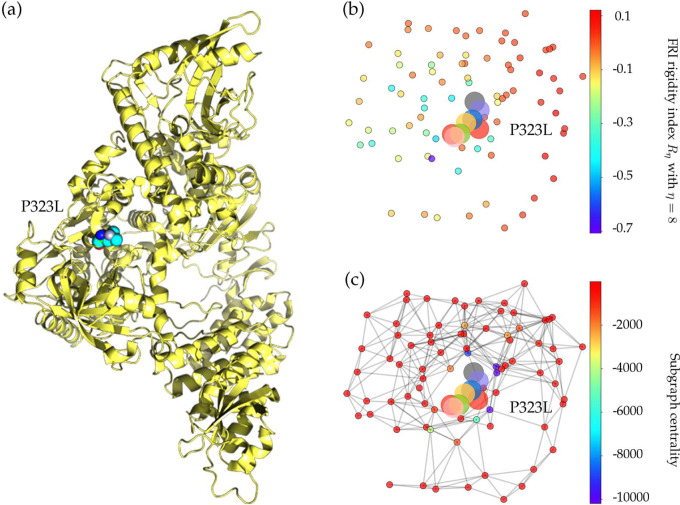
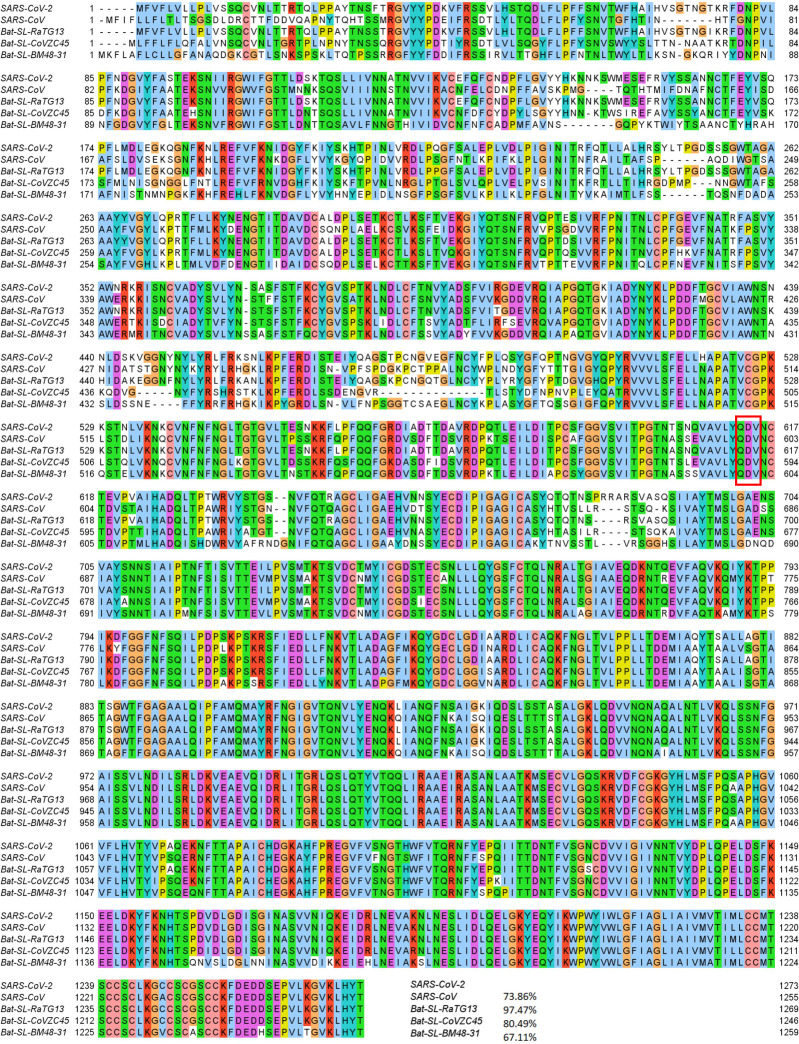
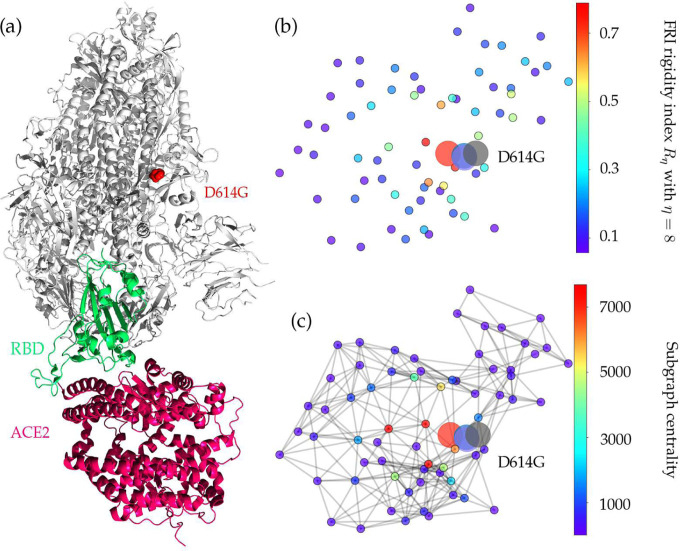
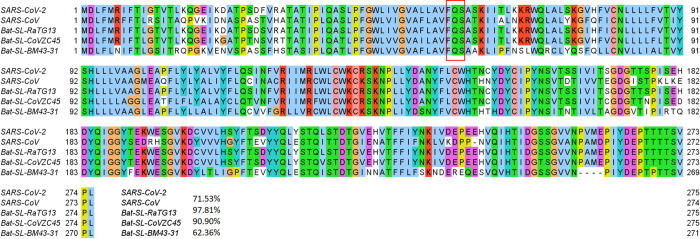
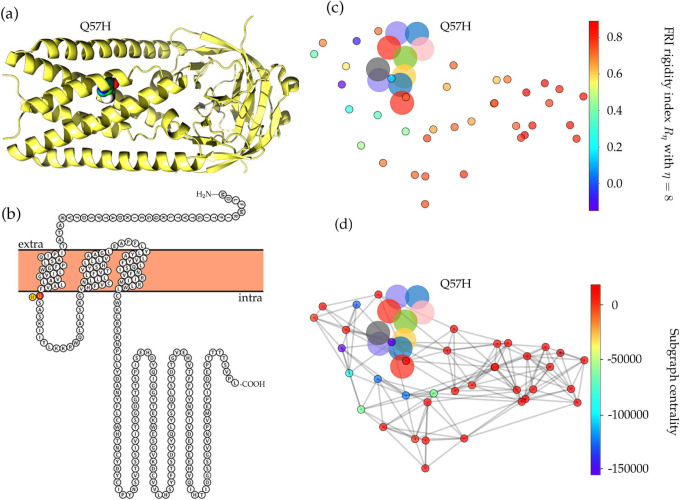
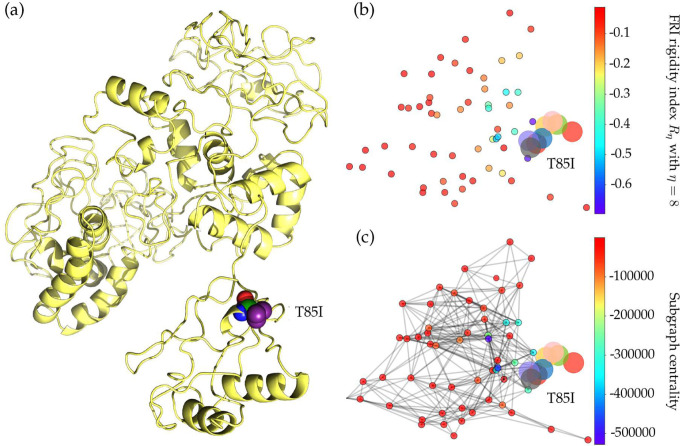
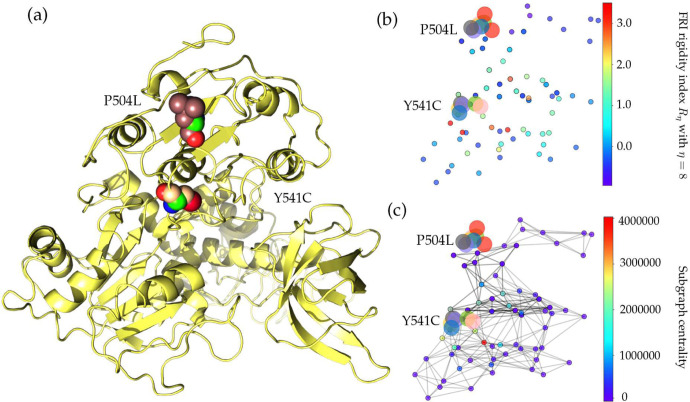
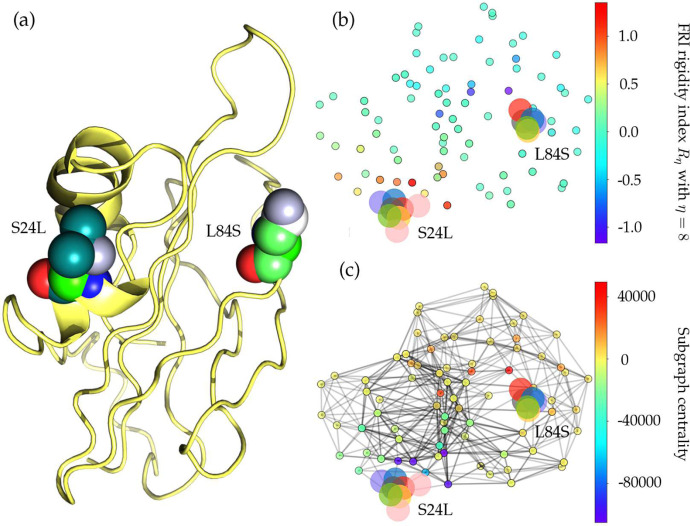
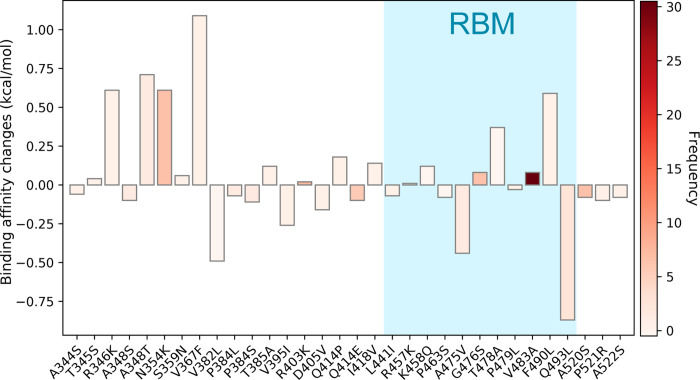
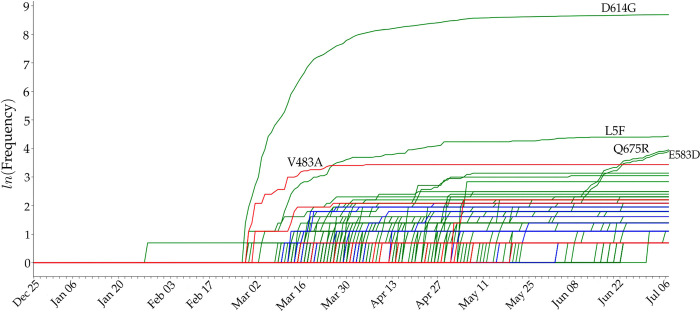
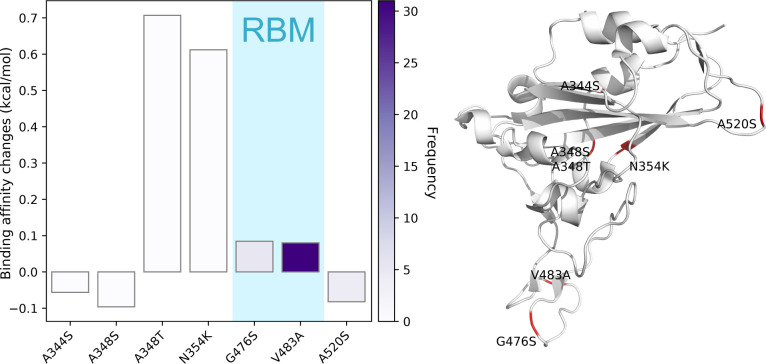
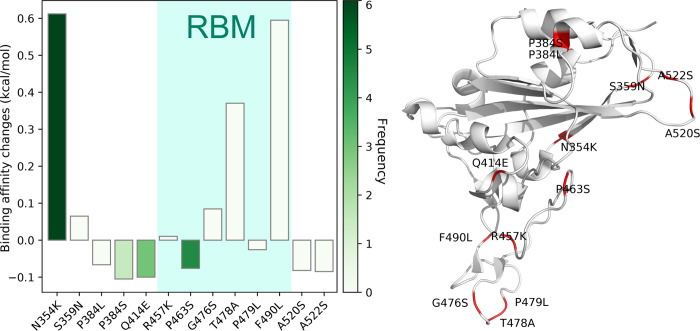
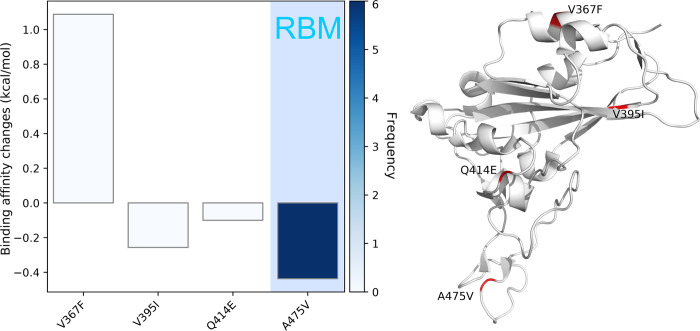
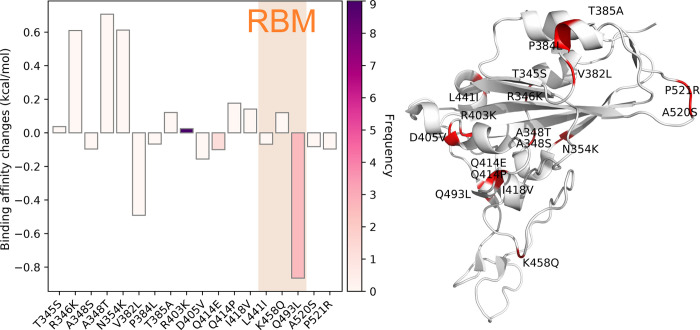
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been mutating since it was first sequenced in early January 2020. The genetic variants have developed into a few distinct clusters with different properties. Since the United States (US) has the highest number of viral infected patients globally, it is essential to understand the US SARS-CoV-2. Using genotyping, sequence-alignment, time-evolution, k-means clustering, protein-folding stability, algebraic topology, and network theory, we reveal that the US SARS-CoV-2 has four substrains and five top US SARS-CoV-2 mutations were first detected in China (2 cases), Singapore (2 cases), and the United Kingdom (1 case). The next three top US SARS-CoV-2 mutations were first detected in the US. These eight top mutations belong to two disconnected groups. The first group consisting of 5 concurrent mutations is prevailing, while the other group with three concurrent mutations gradually fades out. We identify that one of the top mutations, 27964C>T-(S24L) on ORF8, has an unusually strong gender dependence. Based on the analysis of all mutations on the spike protein, we further uncover that three of four US SASR-CoV-2 substrains become more infectious. Our study calls for effective viral control and containing strategies in the US.
Figures



Similar articles
-
Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants.Commun Biol. 2021 Feb 15;4(1):228. doi: 10.1038/s42003-021-01754-6. Commun Biol. 2021. PMID: 33589648 Free PMC article.
-
Transformations, Lineage Comparisons, and Analysis of Down-to-Up Protomer States of Variants of the SARS-CoV-2 Prefusion Spike Protein, Including the UK Variant B.1.1.7.Microbiol Spectr. 2021 Sep 3;9(1):e0003021. doi: 10.1128/Spectrum.00030-21. Epub 2021 Aug 4. Microbiol Spectr. 2021. PMID: 34346753 Free PMC article.
-
Mutations Strengthened SARS-CoV-2 Infectivity.J Mol Biol. 2020 Sep 4;432(19):5212-5226. doi: 10.1016/j.jmb.2020.07.009. Epub 2020 Jul 23. J Mol Biol. 2020. PMID: 32710986 Free PMC article.
-
Emergence, evolution, and vaccine production approaches of SARS-CoV-2 virus: Benefits of getting vaccinated and common questions.Saudi J Biol Sci. 2022 Apr;29(4):1981-1997. doi: 10.1016/j.sjbs.2021.12.020. Epub 2021 Dec 13. Saudi J Biol Sci. 2022. PMID: 34924802 Free PMC article. Review.
-
The Development of SARS-CoV-2 Variants: The Gene Makes the Disease.J Dev Biol. 2021 Dec 15;9(4):58. doi: 10.3390/jdb9040058. J Dev Biol. 2021. PMID: 34940505 Free PMC article. Review.
Cited by
-
Genomic Epidemiology of SARS-CoV-2 Infection During the Initial Pandemic Wave and Association With Disease Severity.JAMA Netw Open. 2021 Apr 1;4(4):e217746. doi: 10.1001/jamanetworkopen.2021.7746. JAMA Netw Open. 2021. PMID: 33900399 Free PMC article.
-
SARS-CoV-2 variant evolution in the United States: High accumulation of viral mutations over time likely through serial Founder Events and mutational bursts.PLoS One. 2021 Jul 23;16(7):e0255169. doi: 10.1371/journal.pone.0255169. eCollection 2021. PLoS One. 2021. PMID: 34297786 Free PMC article.
References
-
- Cang Z. and Wei G.-W.. Analysis and prediction of protein folding energy changes upon mutation by element specific persistent homology. Bioinformatics, 33(22):3549–3557, 2017. - PubMed
-
- Carlsson G.. Topology and data. Bulletin of the American Mathematical Society, 46(2):255–308, 2009.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
