Homozygous deletions implicate non-coding epigenetic marks in Autism spectrum disorder
- PMID: 32820185
- PMCID: PMC7441318
- DOI: 10.1038/s41598-020-70656-0
Homozygous deletions implicate non-coding epigenetic marks in Autism spectrum disorder
Abstract
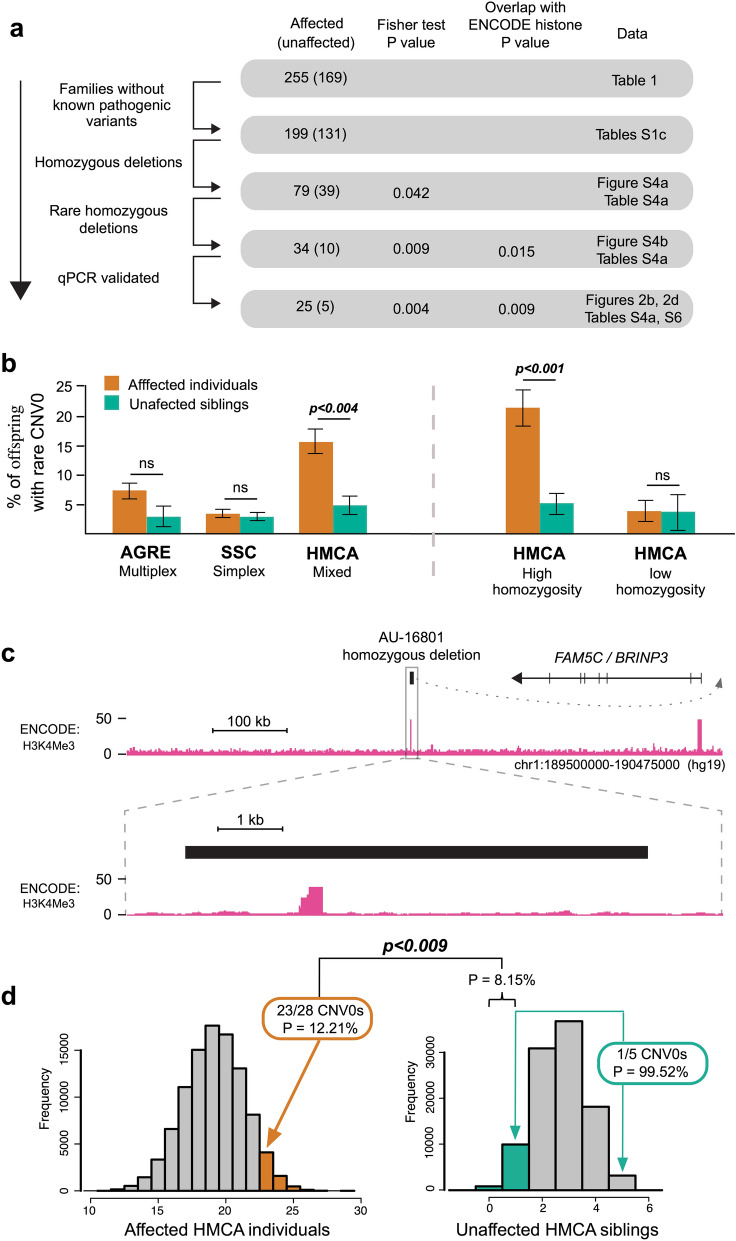
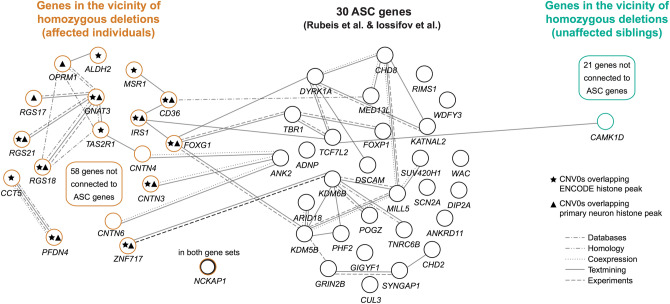
More than 98% of the human genome is made up of non-coding DNA, but techniques to ascertain its contribution to human disease have lagged far behind our understanding of protein coding variations. Autism spectrum disorder (ASD) has been mostly associated with coding variations via de novo single nucleotide variants (SNVs), recessive/homozygous SNVs, or de novo copy number variants (CNVs); however, most ASD cases continue to lack a genetic diagnosis. We analyzed 187 consanguineous ASD families for biallelic CNVs. Recessive deletions were significantly enriched in affected individuals relative to their unaffected siblings (17% versus 4%, p < 0.001). Only a small subset of biallelic deletions were predicted to result in coding exon disruption. In contrast, biallelic deletions in individuals with ASD were enriched for overlap with regulatory regions, with 23/28 CNVs disrupting histone peaks in ENCODE (p < 0.009). Overlap with regulatory regions was further demonstrated by comparisons to the 127-epigenome dataset released by the Roadmap Epigenomics project, with enrichment for enhancers found in primary brain tissue and neuronal progenitor cells. Our results suggest a novel noncoding mechanism of ASD, describe a powerful method to identify important noncoding regions in the human genome, and emphasize the potential significance of gene activation and regulation in cognitive and social function.
Conflict of interest statement
The authors declare no competing interests.
Figures