

Oxidative stress in alcohol-related liver disease
- PMID: 32821333
- PMCID: PMC7407918
- DOI: 10.4254/wjh.v12.i7.332
Oxidative stress in alcohol-related liver disease
Abstract
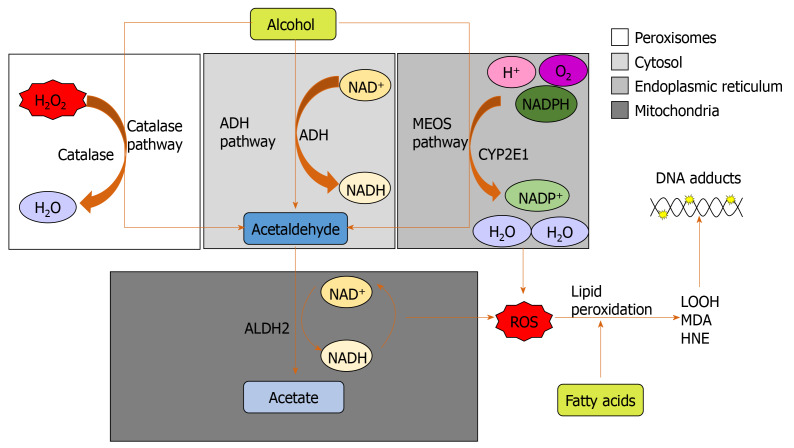
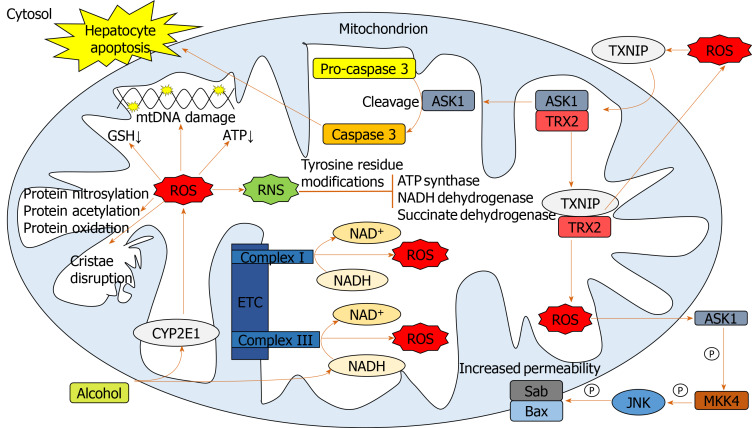
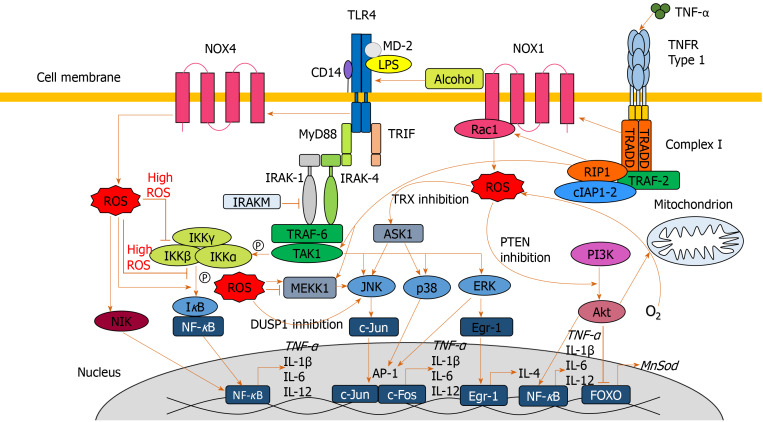
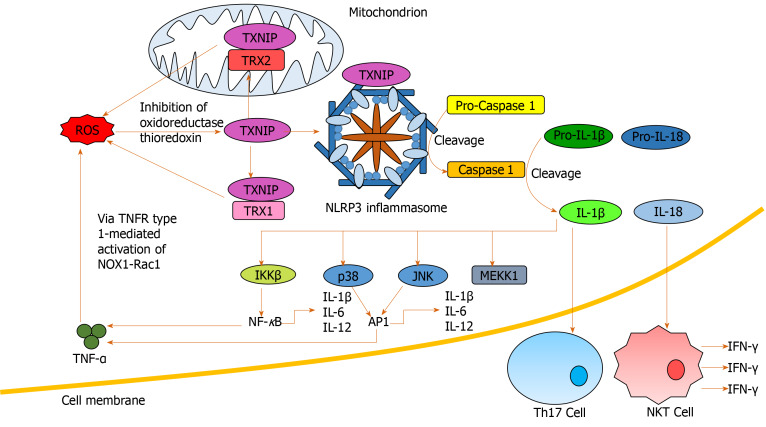
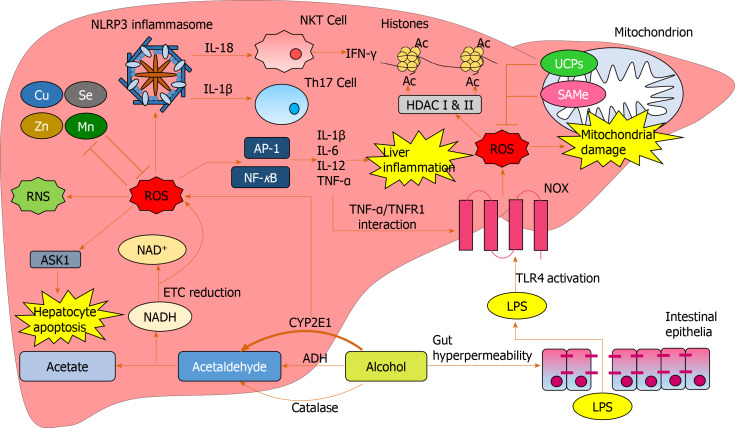
Alcohol consumption is one of the leading causes of the global burden of disease and results in high healthcare and economic costs. Heavy alcohol misuse leads to alcohol-related liver disease, which is responsible for a significant proportion of alcohol-attributable deaths globally. Other than reducing alcohol consumption, there are currently no effective treatments for alcohol-related liver disease. Oxidative stress refers to an imbalance in the production and elimination of reactive oxygen species and antioxidants. It plays important roles in several aspects of alcohol-related liver disease pathogenesis. Here, we review how chronic alcohol use results in oxidative stress through increased metabolism via the cytochrome P450 2E1 system producing reactive oxygen species, acetaldehyde and protein and DNA adducts. These trigger inflammatory signaling pathways within the liver leading to expression of pro-inflammatory mediators causing hepatocyte apoptosis and necrosis. Reactive oxygen species exposure also results in mitochondrial stress within hepatocytes causing structural and functional dysregulation of mitochondria and upregulating apoptotic signaling. There is also evidence that oxidative stress as well as the direct effect of alcohol influences epigenetic regulation. Increased global histone methylation and acetylation and specific histone acetylation inhibits antioxidant responses and promotes expression of key pro-inflammatory genes. This review highlights aspects of the role of oxidative stress in disease pathogenesis that warrant further study including mitochondrial stress and epigenetic regulation. Improved understanding of these processes may identify novel targets for therapy.
Keywords: Alcohol-related liver disease; Alcoholic hepatitis; Antioxidants; Epigenetics; Mitochondrial stress; Oxidative stress; Reactive oxygen species.
©The Author(s) 2020. Published by Baishideng Publishing Group Inc. All rights reserved.
Figures
References
-
- Pimpin L, Cortez-Pinto H, Negro F, Corbould E, Lazarus JV, Webber L, Sheron N EASL HEPAHEALTH Steering Committee. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J Hepatol. 2018;69:718–735. - PubMed
-
- Bhattacharya A. Which cost of alcohol? What should we compare it against? Addiction. 2017;112:559–565. - PubMed
-
- Anderson P, Baumberg B. Alcohol in Europe-Public Health Perspective: Report summary. Drugs Educ Prev Policy. 2006;13:483–488.
Publication types
LinkOut - more resources
Full Text Sources
