

Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic
- PMID: 32824573
- PMCID: PMC7464704
- DOI: 10.3390/genes11080949
Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic
Abstract
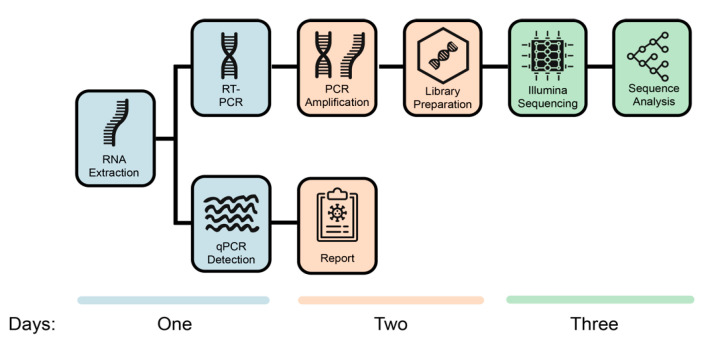
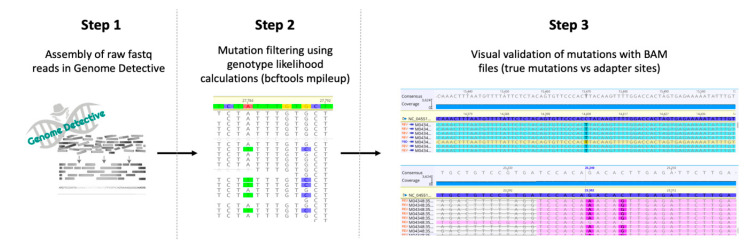
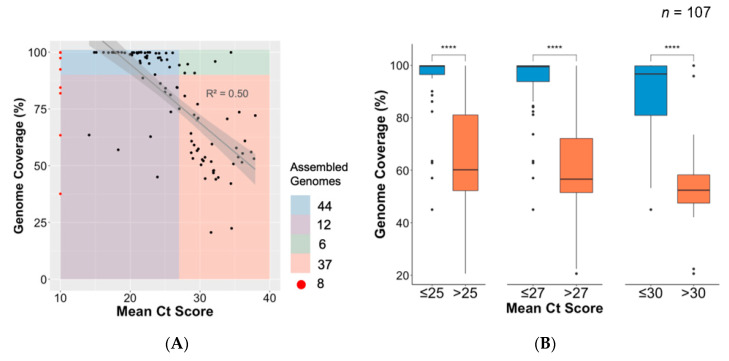
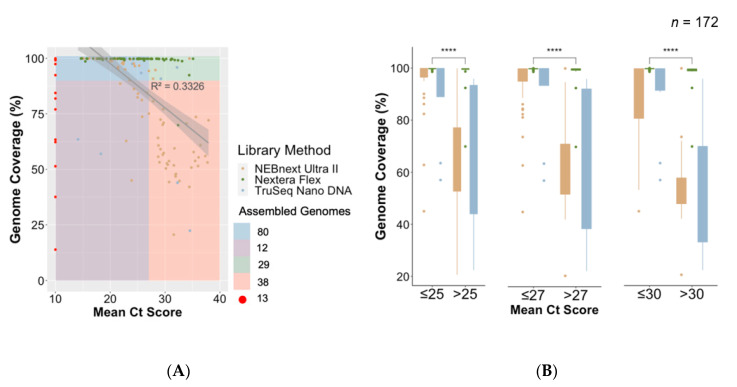
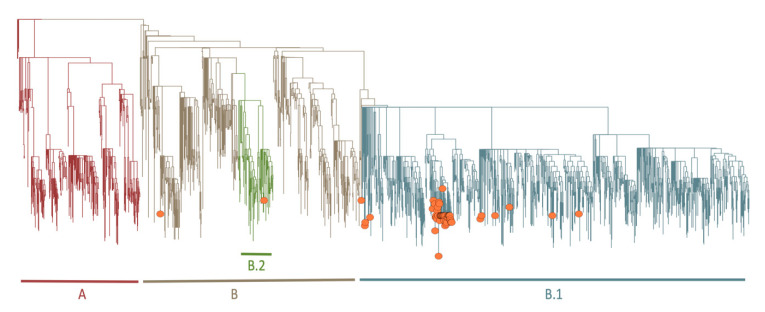
The COVID-19 pandemic has spread very fast around the world. A few days after the first detected case in South Africa, an infection started in a large hospital outbreak in Durban, KwaZulu-Natal (KZN). Phylogenetic analysis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genomes can be used to trace the path of transmission within a hospital. It can also identify the source of the outbreak and provide lessons to improve infection prevention and control strategies. This manuscript outlines the obstacles encountered in order to genotype SARS-CoV-2 in near-real time during an urgent outbreak investigation. This included problems with the length of the original genotyping protocol, unavailability of reagents, and sample degradation and storage. Despite this, three different library preparation methods for Illumina sequencing were set up, and the hands-on library preparation time was decreased from twelve to three hours, which enabled the outbreak investigation to be completed in just a few weeks. Furthermore, the new protocols increased the success rate of sequencing whole viral genomes. A simple bioinformatics workflow for the assembly of high-quality genomes in near-real time was also fine-tuned. In order to allow other laboratories to learn from our experience, all of the library preparation and bioinformatics protocols are publicly available at protocols.io and distributed to other laboratories of the Network for Genomics Surveillance in South Africa (NGS-SA) consortium.
Keywords: COVID-19; Illumina; SARS-CoV2; bioinformatics; protocols; sequencing.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
