

TLR3-Dependent Activation of TLR2 Endogenous Ligands via the MyD88 Signaling Pathway Augments the Innate Immune Response
- PMID: 32824595
- PMCID: PMC7464415
- DOI: 10.3390/cells9081910
TLR3-Dependent Activation of TLR2 Endogenous Ligands via the MyD88 Signaling Pathway Augments the Innate Immune Response
Abstract
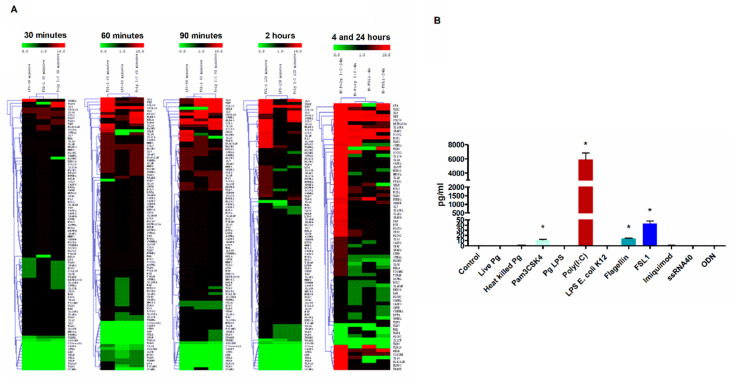
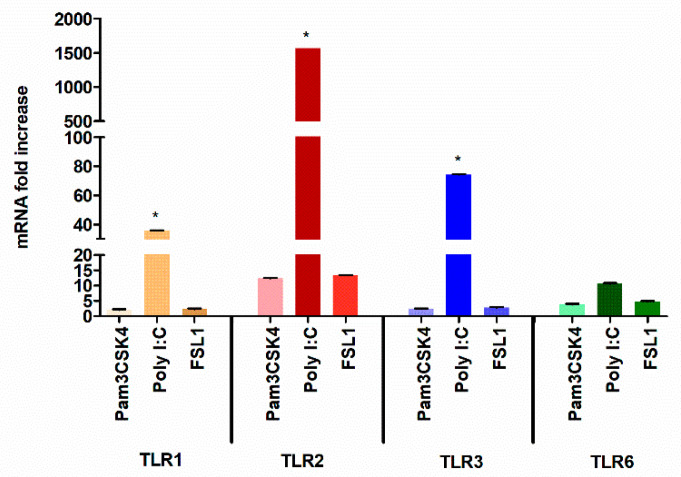
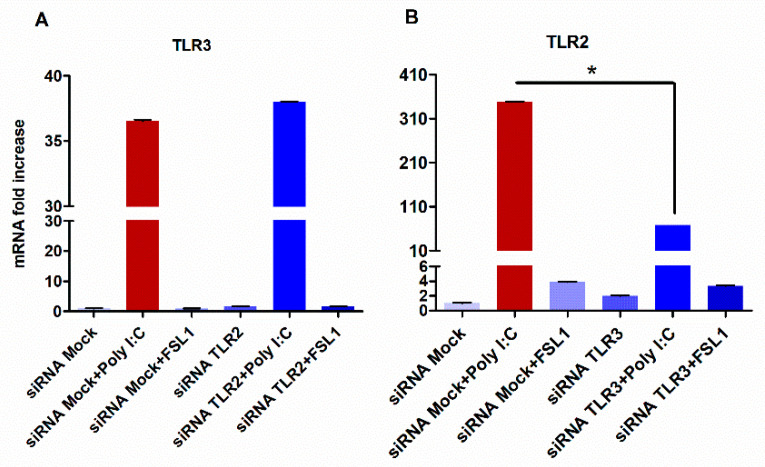
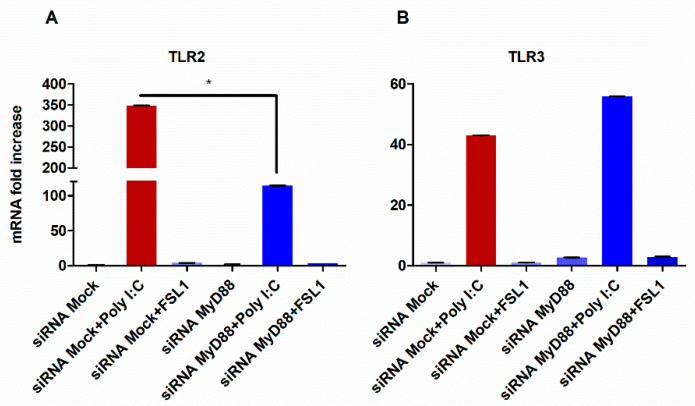
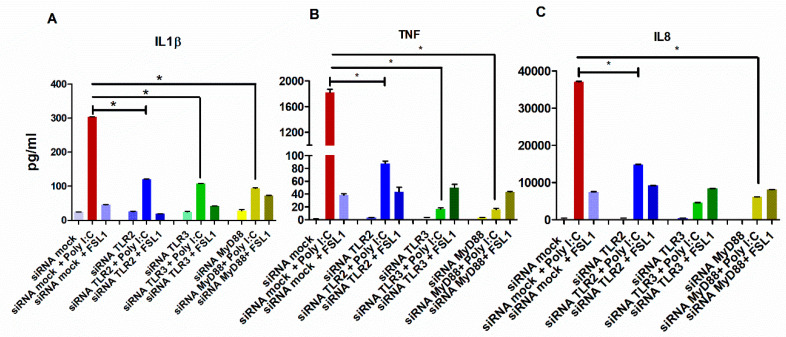
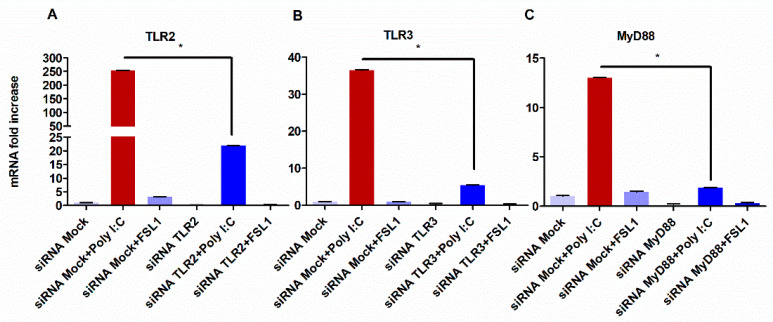
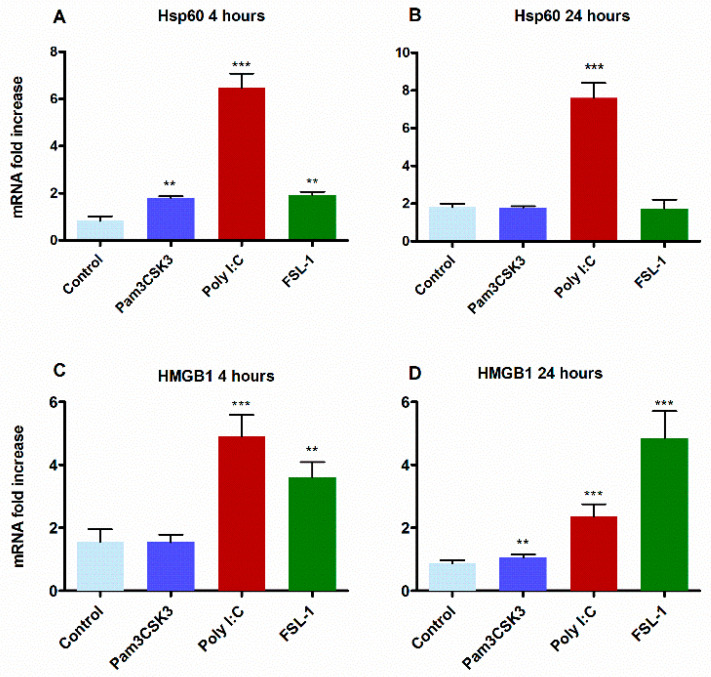
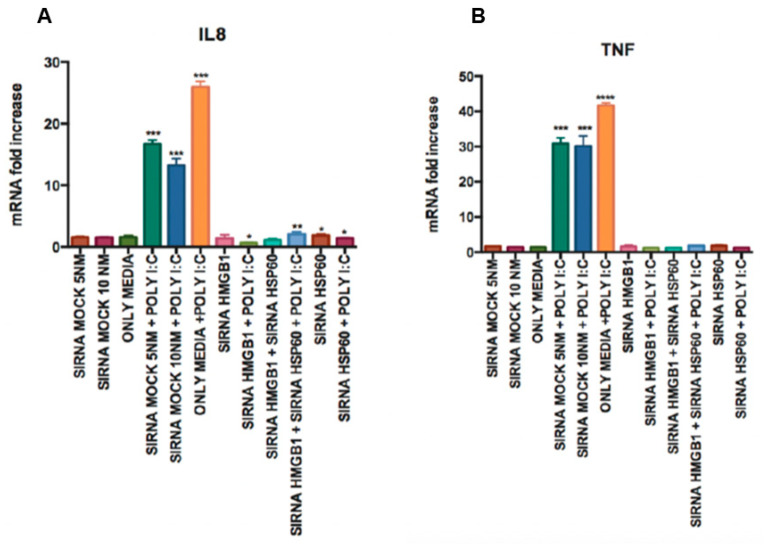
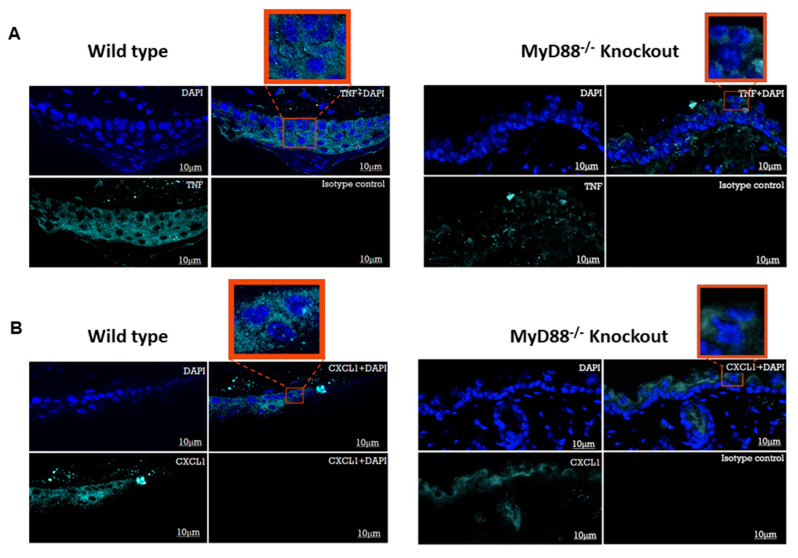
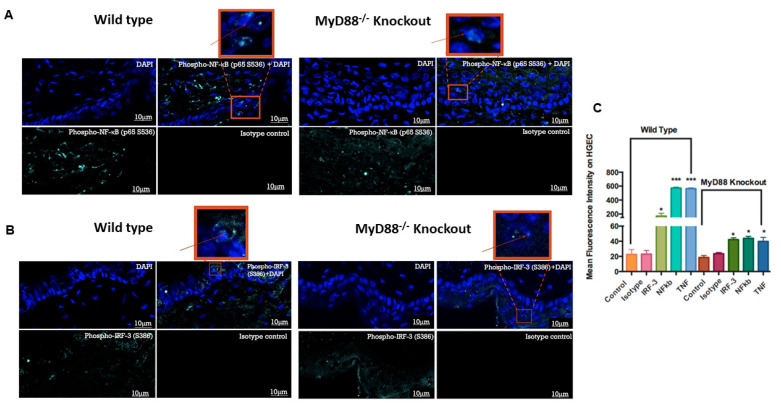
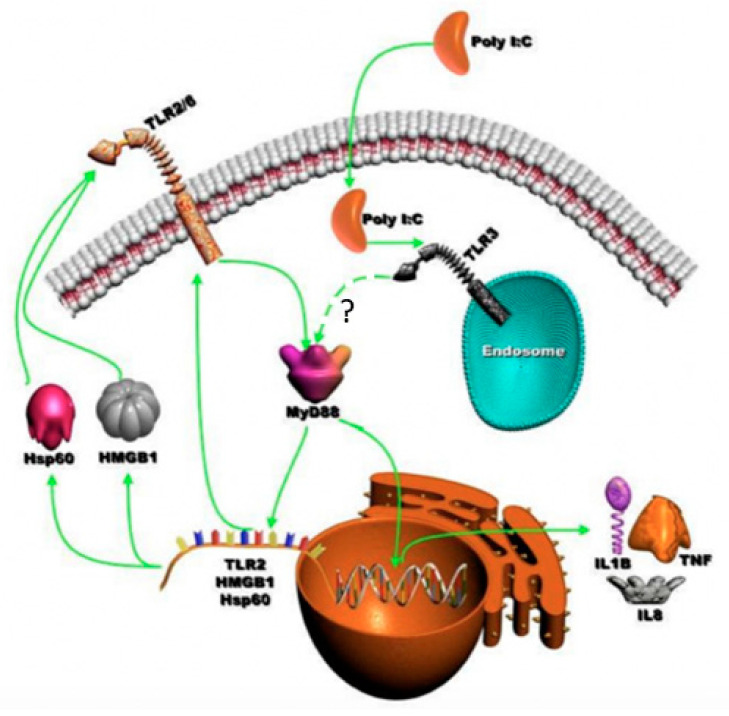
The role of the adaptor molecule MyD88 is thought to be independent of Toll-like receptor 3 (TLR3) signaling. In this report, we demonstrate a previously unknown role of MyD88 in TLR3 signaling in inducing endogenous ligands of TLR2 to elicit innate immune responses. Of the various TLR ligands examined, the TLR3-specific ligand polyinosinic:polycytidylic acid (poly I:C), significantly induced TNF production and the upregulation of other TLR transcripts, in particular, TLR2. Accordingly, TLR3 stimulation also led to a significant upregulation of endogenous TLR2 ligands mainly, HMGB1 and Hsp60. By contrast, the silencing of TLR3 significantly downregulated MyD88 and TLR2 gene expression and pro-inflammatory IL1β, TNF, and IL8 secretion. The silencing of MyD88 similarly led to the downregulation of TLR2, IL1β, TNF and IL8, thus suggesting MyD88 to somehow act downstream of TLR3. Corroborating in vitro data, Myd88-/- knockout mice downregulated TNF, CXCL1; and phospho-p65 and phospho-IRF3 nuclear localization, upon poly I:C treatment in a mouse model of skin infection. Taken together, we identified a previously unknown role for MyD88 in the TLR3 signaling pathway, underlying the importance of TLRs and adapter protein interplay in modulating endogenous TLR ligands culminating in pro-inflammatory cytokine regulation.
Keywords: HMGB1; Hsp60; MyD88; TLR3; human gingival epithelial cells; pro-inflammatory cytokine.
Conflict of interest statement
Authors declare no conflict of interest.
Figures
Similar articles
-
Recognition of Candida albicans by gingival fibroblasts: The role of TLR2, TLR4/CD14, and MyD88.Cytokine. 2018 Jun;106:67-75. doi: 10.1016/j.cyto.2017.10.013. Epub 2017 Nov 9. Cytokine. 2018. PMID: 29128406 Free PMC article.
-
TLR3-mediated IFN-β gene induction is negatively regulated by the TLR adaptor MyD88 adaptor-like.Eur J Immunol. 2010 Nov;40(11):3150-60. doi: 10.1002/eji.201040547. Epub 2010 Oct 19. Eur J Immunol. 2010. PMID: 20957750
-
Trif is not required for immune complex glomerulonephritis: dying cells activate mesangial cells via Tlr2/Myd88 rather than Tlr3/Trif.Am J Physiol Renal Physiol. 2009 Apr;296(4):F867-74. doi: 10.1152/ajprenal.90213.2008. Epub 2009 Jan 21. Am J Physiol Renal Physiol. 2009. PMID: 19158348
-
Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and Toll-like receptor/MyD88 pathways.Cancer Sci. 2016 Apr;107(4):391-7. doi: 10.1111/cas.12901. Epub 2016 Mar 18. Cancer Sci. 2016. PMID: 27079437 Free PMC article. Review.
-
Impact of Toll-Like Receptors (TLRs) and TLR Signaling Proteins in Trigeminal Ganglia Impairing Herpes Simplex Virus 1 (HSV-1) Progression to Encephalitis: Insights from Mouse Models.Front Biosci (Landmark Ed). 2024 Mar 14;29(3):102. doi: 10.31083/j.fbl2903102. Front Biosci (Landmark Ed). 2024. PMID: 38538263 Review.
Cited by
-
Myeloid MyD88 Mediates Macrophage Infiltration and Activation in Ang II-Induced Cardiac Hypertrophy.J Cell Mol Med. 2025 Jul;29(14):e70733. doi: 10.1111/jcmm.70733. J Cell Mol Med. 2025. PMID: 40682481 Free PMC article.
-
A "Drug-Dependent" Immune System Can Compromise Protection against Infection: The Relationships between Psychostimulants and HIV.Viruses. 2021 Apr 21;13(5):722. doi: 10.3390/v13050722. Viruses. 2021. PMID: 33919273 Free PMC article. Review.
-
Antineoplastic with DNA fragmentation assay and anti-oxidant, anti-inflammatory with gene expression activity of Lactobacillus plantarum isolated from local Egyptian milk products.BMC Microbiol. 2024 Oct 29;24(1):443. doi: 10.1186/s12866-024-03576-y. BMC Microbiol. 2024. PMID: 39472774 Free PMC article.
-
Unraveling cell death mechanisms in traumatic brain injury: dynamic roles of ferroptosis and necroptosis.Mol Biol Rep. 2025 Apr 10;52(1):381. doi: 10.1007/s11033-025-10489-0. Mol Biol Rep. 2025. PMID: 40208458 Review.
-
Immunological Strategies in Gastric Cancer: How Toll-like Receptors 2, -3, -4, and -9 on Monocytes and Dendritic Cells Depend on Patient Factors?Cells. 2024 Oct 16;13(20):1708. doi: 10.3390/cells13201708. Cells. 2024. PMID: 39451226 Free PMC article.
References
-
- Alkanani A.K., Hara N., Lien E., Ir D., Kotter C.V., Robertson C.E., Wagner B.D., Frank D.N., Zipris D. Induction of diabetes in the RIP-B7.1 mouse model is critically dependent on TLR3 and MyD88 pathways and is associated with alterations in the intestinal microbiome. Diabetes. 2014;63:619–631. doi: 10.2337/db13-1007. - DOI - PubMed
-
- Liang L., Tan X., Zhou Q., Tian Y., Kijlstra A., Yang P. TLR3 and TLR4 But not TLR2 are involved in vogt-koyanagi- harada disease by triggering proinflammatory cytokines production through promoting the production of mitochondrial reactive oxygen species. Curr. Mol. Med. 2015;15:529–542. doi: 10.2174/1566524015666150731095611. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
