

Hierarchical natural move Monte Carlo refines flexible RNA structures into cryo-EM densities
- PMID: 32826323
- PMCID: PMC7668250
- DOI: 10.1261/rna.071100.119
Hierarchical natural move Monte Carlo refines flexible RNA structures into cryo-EM densities
Abstract
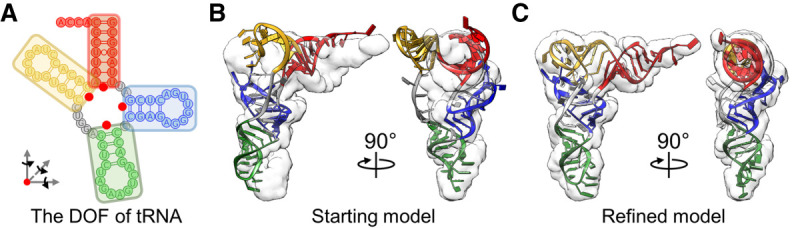
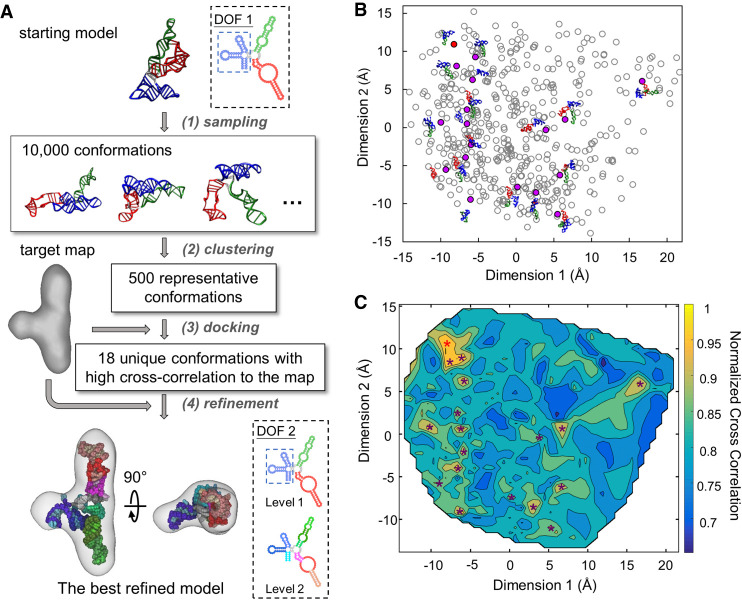
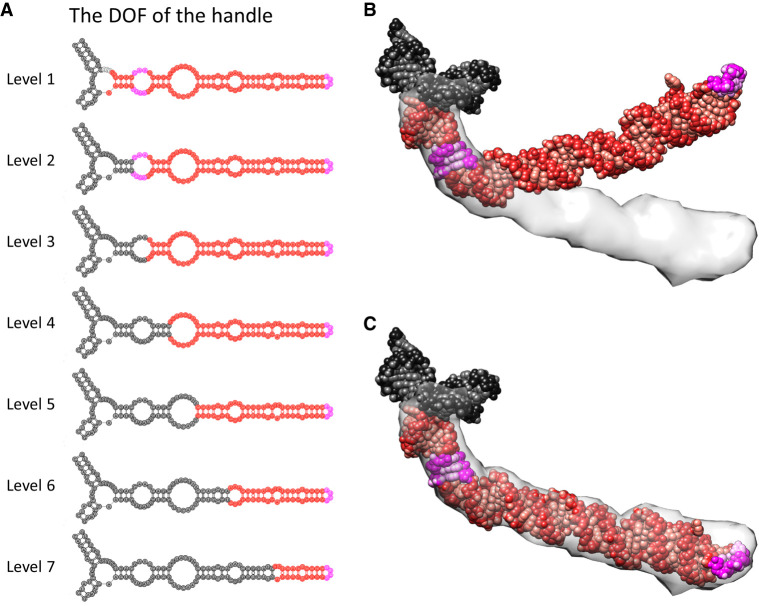
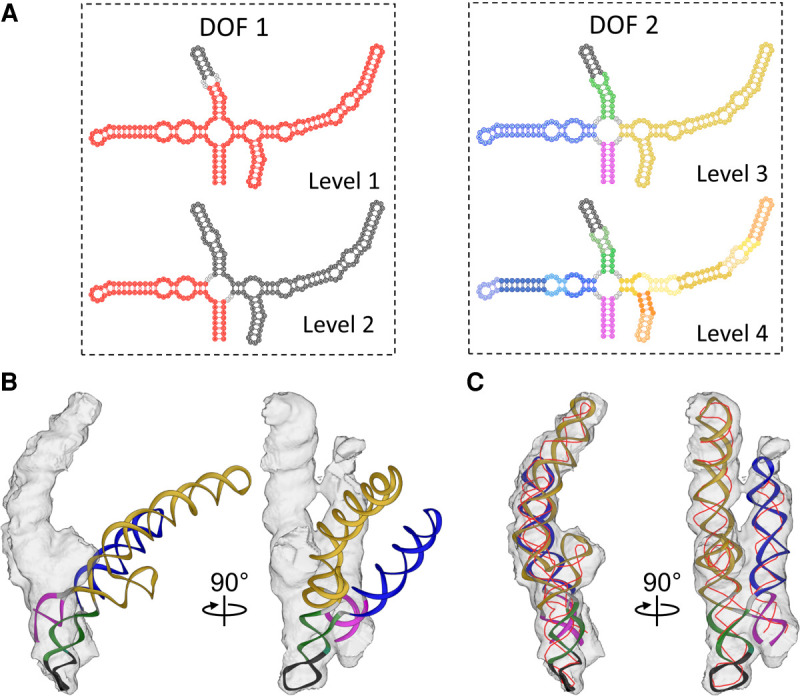
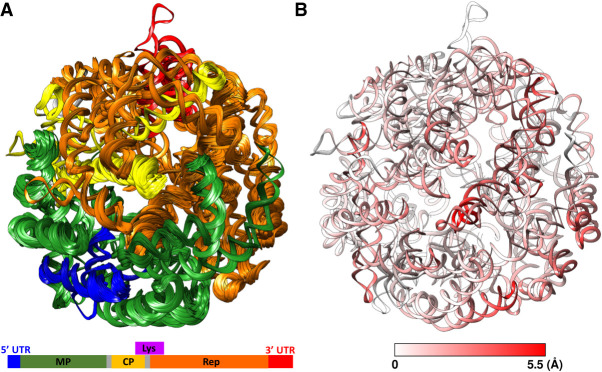
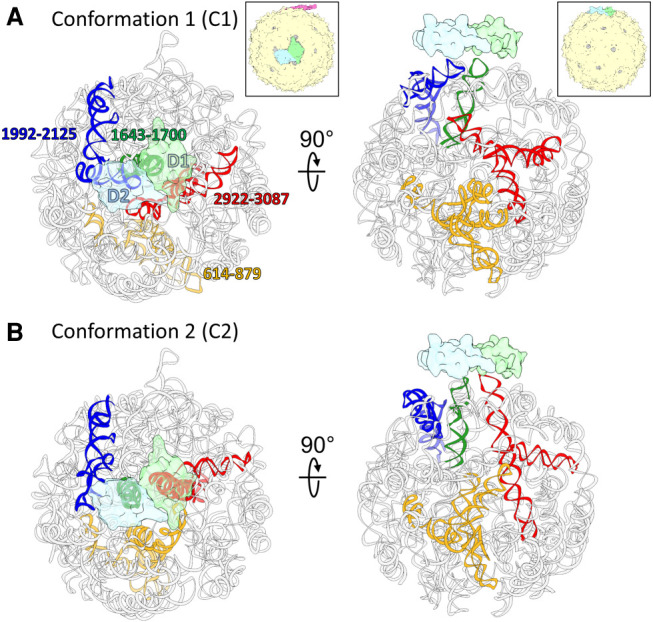
Ribonucleic acids (RNAs) play essential roles in living cells. Many of them fold into defined three-dimensional (3D) structures to perform functions. Recent advances in single-particle cryo-electron microscopy (cryo-EM) have enabled structure determinations of RNA to atomic resolutions. However, most RNA molecules are structurally flexible, limiting the resolution of their structures solved by cryo-EM. In modeling these molecules, several computational methods are limited by the requirement of massive computational resources and/or the low efficiency in exploring large-scale structural variations. Here we use hierarchical natural move Monte Carlo (HNMMC), which takes advantage of collective motions for groups of nucleic acid residues, to refine RNA structures into their cryo-EM maps, preserving atomic details in the models. After validating the method on a simulated density map of tRNA, we applied it to objectively obtain the model of the folding intermediate for the specificity domain of ribonuclease P from Bacillus subtilis and refine a flexible ribosomal RNA (rRNA) expansion segment from the Mycobacterium tuberculosis (Mtb) ribosome in different conformational states. Finally, we used HNMMC to model atomic details and flexibility for two distinct conformations of the complete genomic RNA (gRNA) inside MS2, a single-stranded RNA virus, revealing multiple pathways for its capsid assembly.
Keywords: RNA folding; RNA virus; conformational sampling; molecular motions; structural modeling.
© 2020 Chang et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases