

Dual effects of thyroid hormone on neurons and neurogenesis in traumatic brain injury
- PMID: 32826870
- PMCID: PMC7442821
- DOI: 10.1038/s41419-020-02836-9
Dual effects of thyroid hormone on neurons and neurogenesis in traumatic brain injury
Abstract
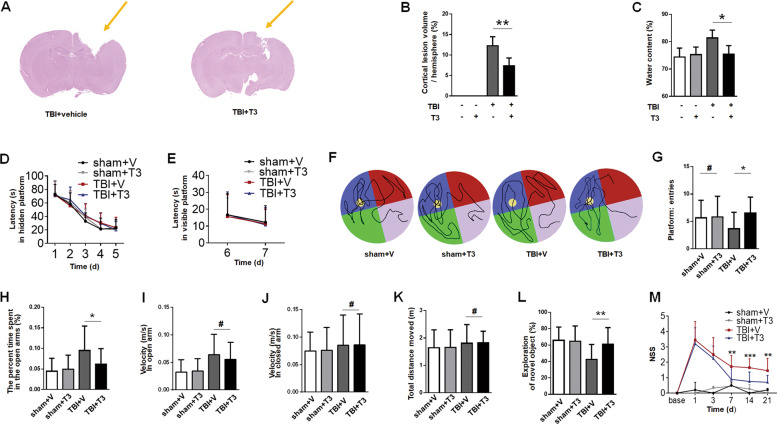
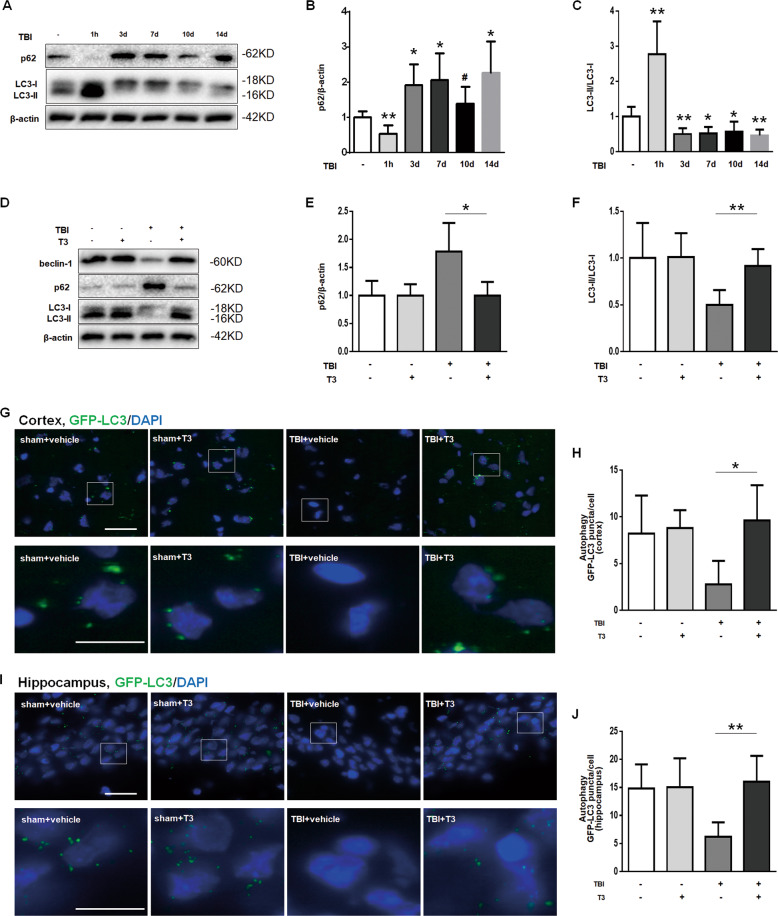
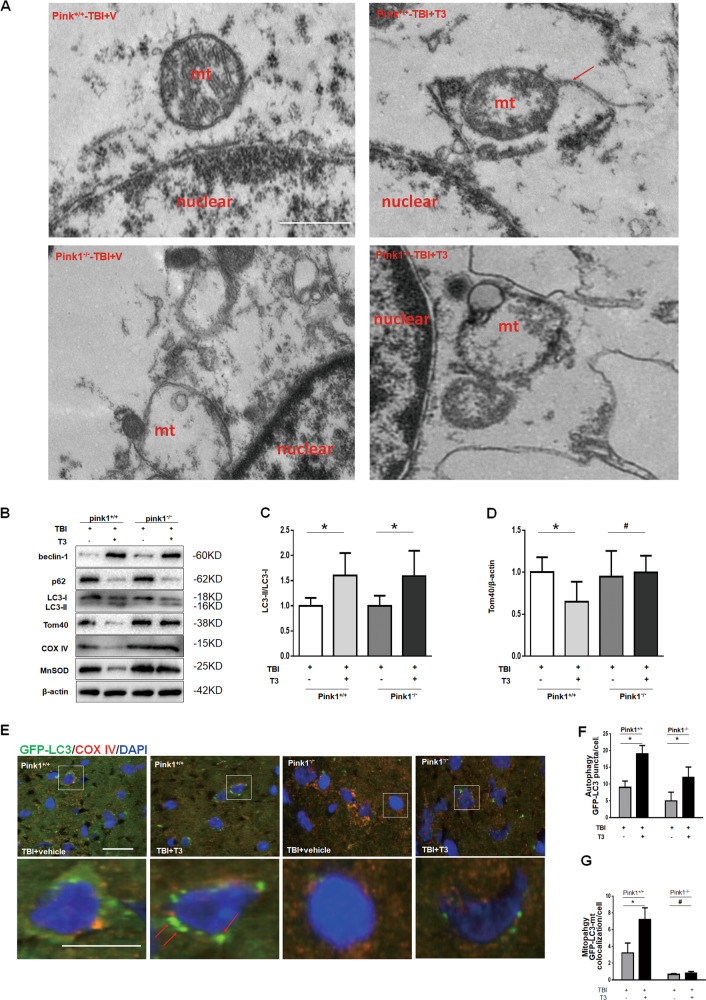
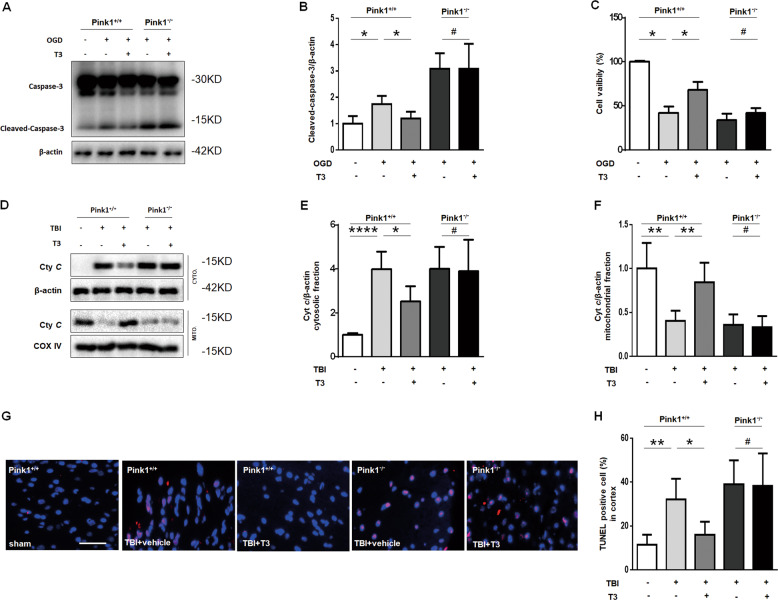
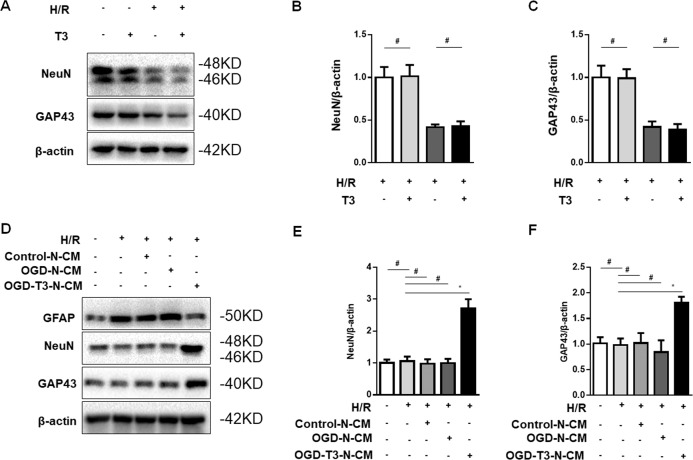
Thyroid hormone (TH) plays a crucial role in neurodevelopment, but its function and specific mechanisms remain unclear after traumatic brain injury (TBI). Here we found that treatment with triiodothyronine (T3) ameliorated the progression of neurological deficits in mice subjected to TBI. The data showed that T3 reduced neural death and promoted the elimination of damaged mitochondria via mitophagy. However, T3 did not prevent TBI-induced cell death in phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (Pink1) knockout mice suggesting the involvement of mitophagy. Moreover, we also found that T3 promoted neurogenesis via crosstalk between mature neurons and neural stem cells (NSCs) after TBI. In neuron cultures undergoing oxygen and glucose deprivation (OGD), conditioned neuron culture medium collected after T3 treatment enhanced the in vitro differentiation of NSCs into mature neurons, a process in which mitophagy was required. Taken together, these data suggested that T3 treatment could provide a therapeutic approach for TBI by preventing neuronal death via mitophagy and promoting neurogenesis via neuron-NSC crosstalk.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
