

CBFB-MYH11 fusion neoantigen enables T cell recognition and killing of acute myeloid leukemia
- PMID: 32831296
- PMCID: PMC7524498
- DOI: 10.1172/JCI137723
CBFB-MYH11 fusion neoantigen enables T cell recognition and killing of acute myeloid leukemia
Abstract
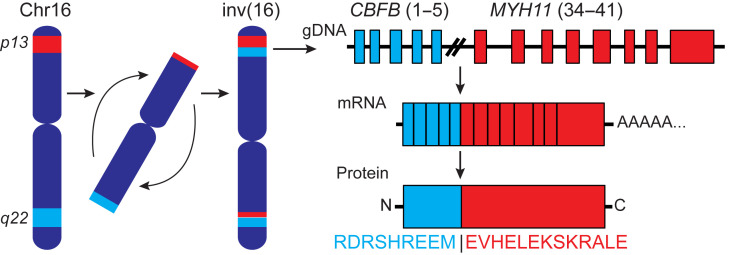
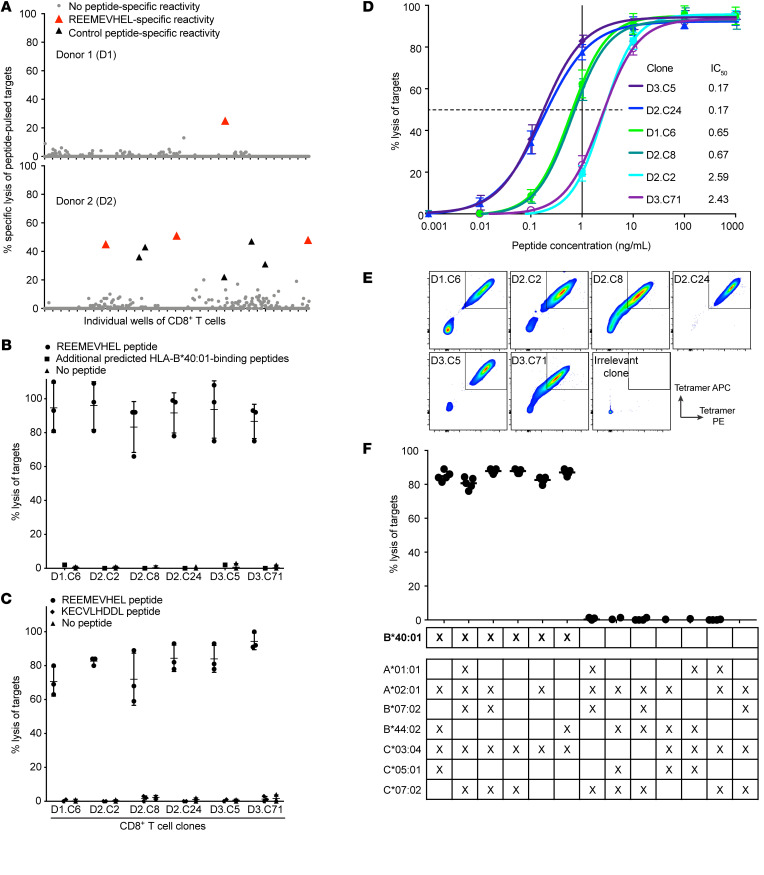
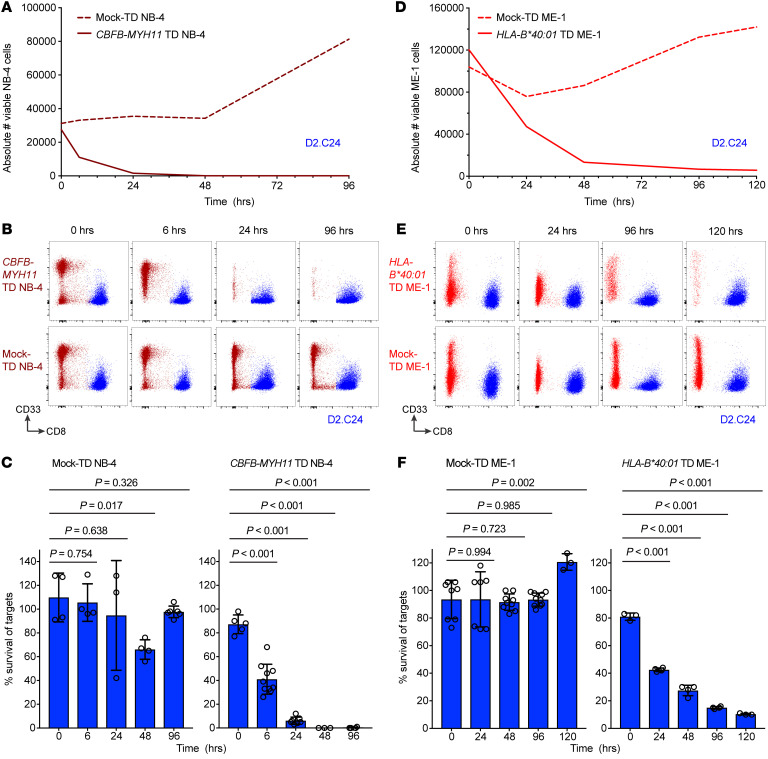
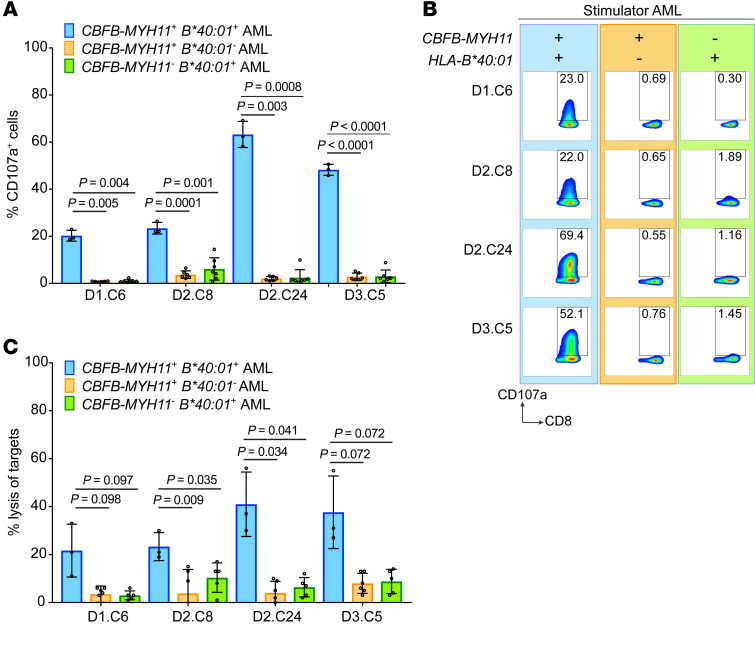
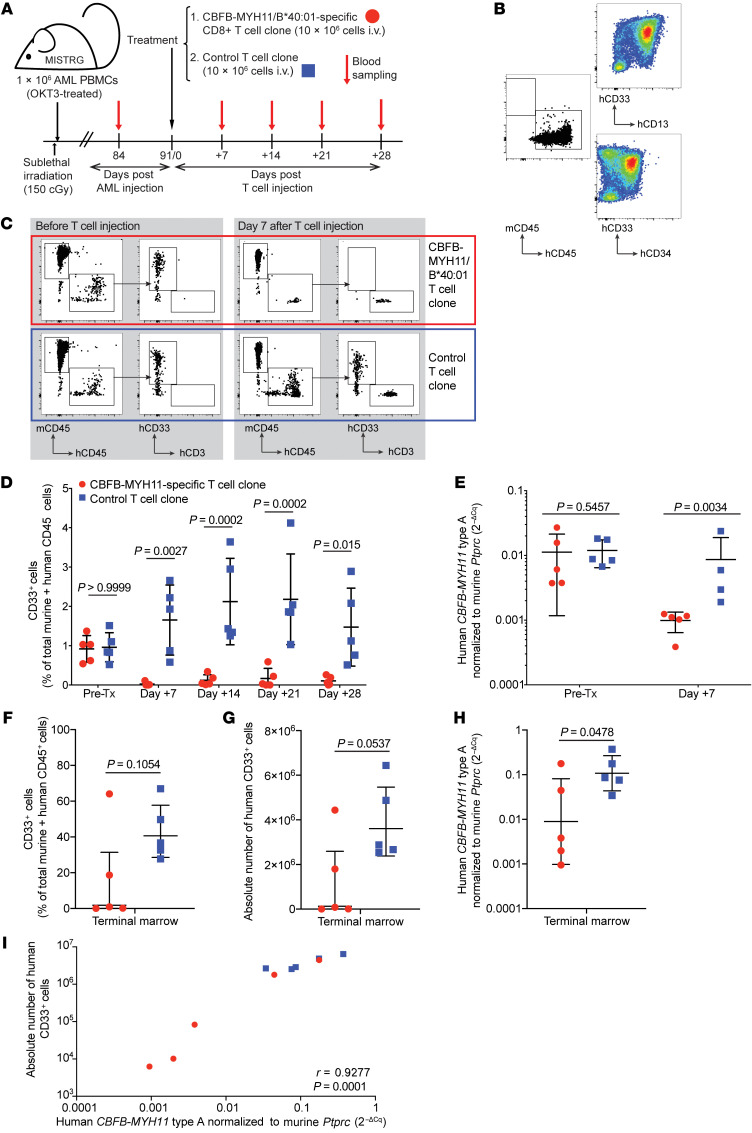
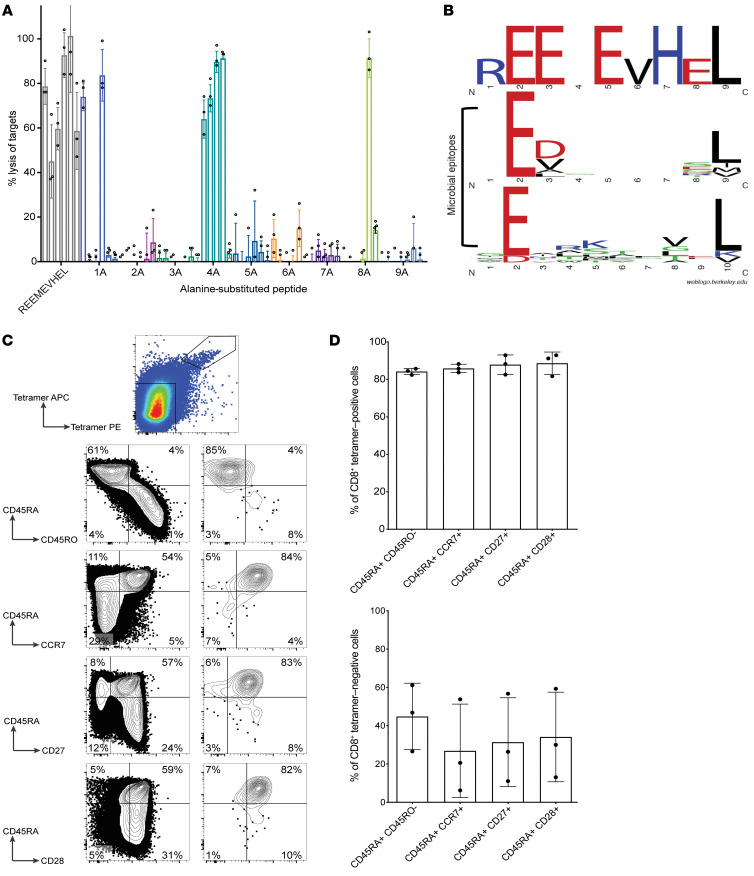
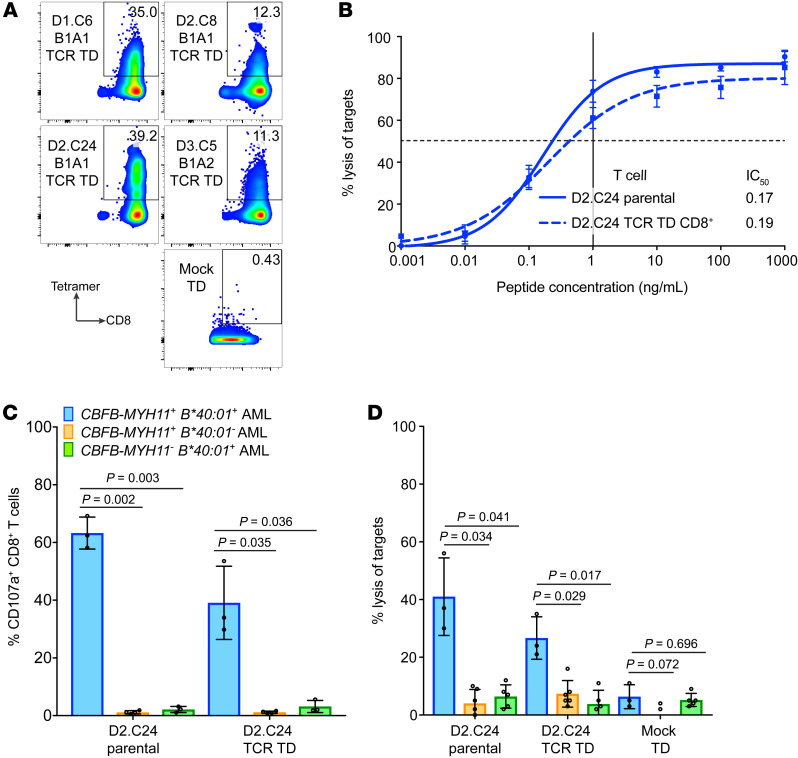
Proteins created from recurrent fusion genes like CBFB-MYH11 are prevalent in acute myeloid leukemia (AML), often necessary for leukemogenesis, persistent throughout the disease course, and highly leukemia specific, making them attractive neoantigen targets for immunotherapy. A nonameric peptide derived from a prevalent CBFB-MYH11 fusion protein was found to be immunogenic in HLA-B*40:01+ donors. High-avidity CD8+ T cell clones isolated from healthy donors killed CBFB-MYH11+ HLA-B*40:01+ AML cell lines and primary human AML samples in vitro. CBFB-MYH11-specific T cells also controlled CBFB-MYH11+ HLA-B*40:01+ AML in vivo in a patient-derived murine xenograft model. High-avidity CBFB-MYH11 epitope-specific T cell receptors (TCRs) transduced into CD8+ T cells conferred antileukemic activity in vitro. Our data indicate that the CBFB-MYH11 fusion neoantigen is naturally presented on AML blasts and enables T cell recognition and killing of AML. We provide proof of principle for immunologically targeting AML-initiating fusions and demonstrate that targeting neoantigens has clinical relevance even in low-mutational frequency cancers like fusion-driven AML. This work also represents a first critical step toward the development of TCR T cell immunotherapy targeting fusion gene-driven AML.
Keywords: Cancer immunotherapy; Immunology; Leukemias; Oncology; T cells.
Conflict of interest statement
Figures
Comment on
-
Inactivation of paracellular cation-selective claudin-2 channels attenuates immune-mediated experimental colitis in mice.J Clin Invest. 2020 Oct 1;130(10):5197-5208. doi: 10.1172/JCI138697. J Clin Invest. 2020. PMID: 32516134 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
