

Computational Modeling Predicts Immuno-Mechanical Mechanisms of Maladaptive Aortic Remodeling in Hypertension
- PMID: 32831391
- PMCID: PMC7437922
- DOI: 10.1016/j.ijengsci.2019.05.014
Computational Modeling Predicts Immuno-Mechanical Mechanisms of Maladaptive Aortic Remodeling in Hypertension
Abstract
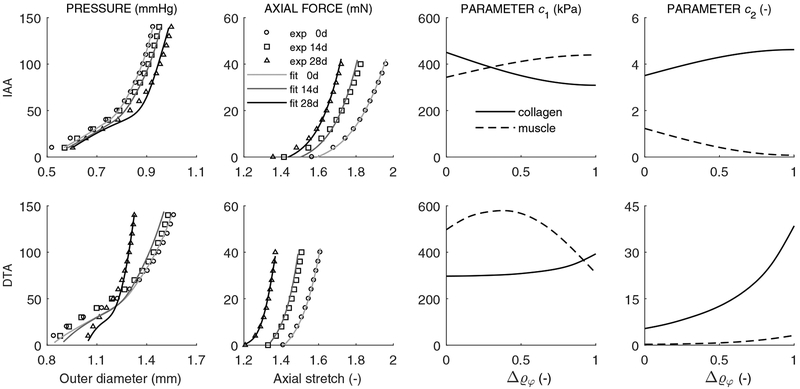
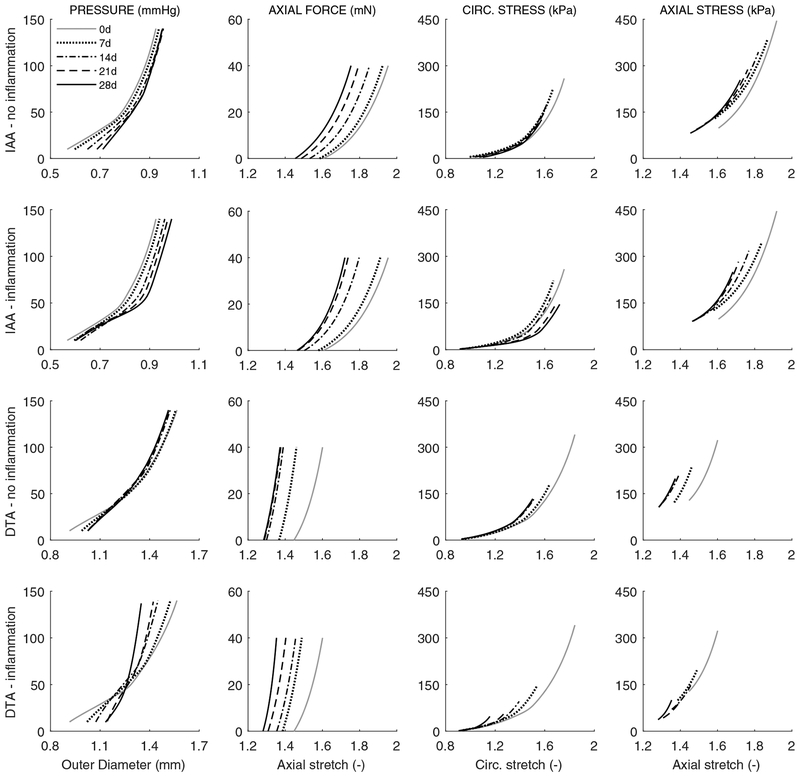
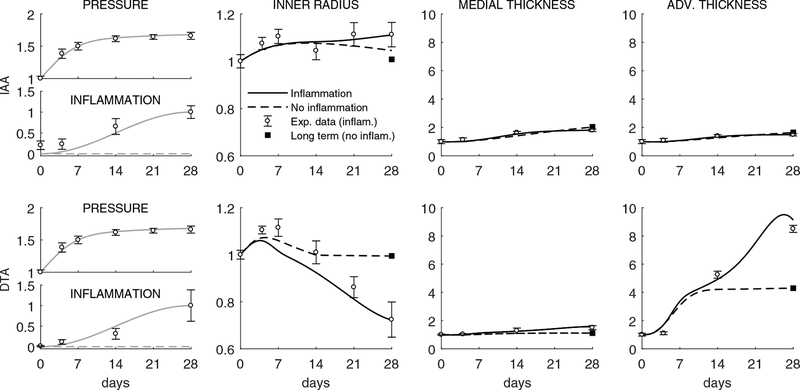
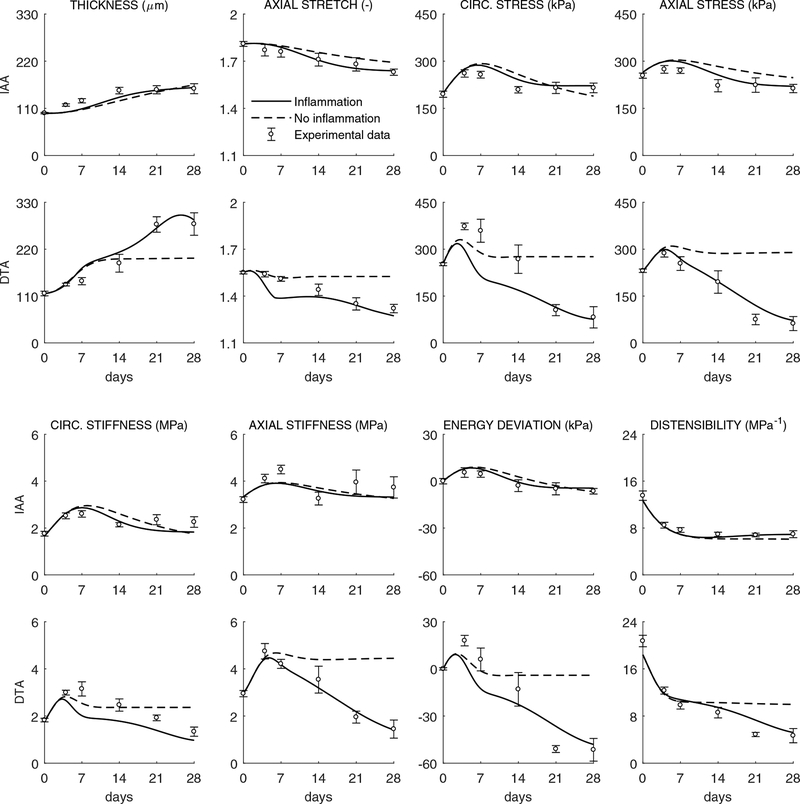
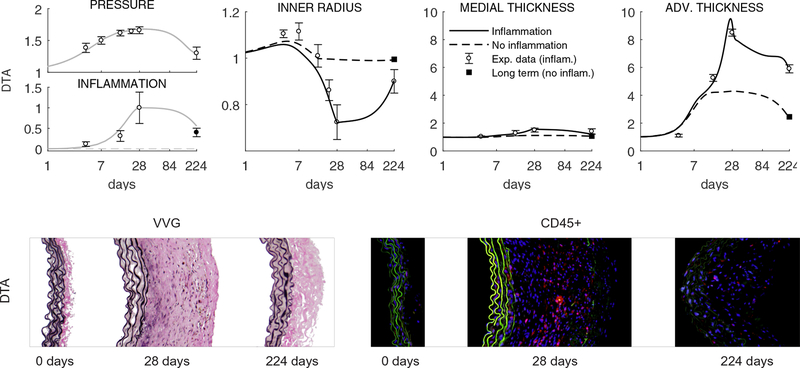
Uncontrolled hypertension is a major risk factor for myriad cardiovascular diseases. Among its many effects, hypertension increases central artery stiffness which in turn is both an initiator and indicator of disease. Despite extensive clinical, animal, and basic science studies, the biochemomechanical mechanisms by which hypertension drives aortic stiffening remain unclear. In this paper, we show that a new computational model of aortic growth and remodeling can capture differential effects of induced hypertension on the thoracic and abdominal aorta in a common mouse model of disease. Because the simulations treat the aortic wall as a constrained mixture of different constituents having different material properties and rates of turnover, one can gain increased insight into underlying constituent-level mechanisms of aortic remodeling. Model results suggest that the aorta can mechano-adapt locally to blood pressure elevation in the absence of marked inflammation, but large increases in inflammation drive a persistent maladaptive phenotype characterized primarily by adventitial fibrosis. Moreover, this fibrosis appears to occur via a marked increase in the rate of deposition of collagen having different material properties in the absence of a compensatory increase in the rate of matrix degradation. Controlling inflammation thus appears to be key to reducing fibrosis, but therapeutic strategies should not compromise the proteolytic activity of the wall that is essential to mechanical homeostasis.
Keywords: aorta; blood pressure; constrained mixture; inflammation; wall stress.
Conflict of interest statement
Conflict of Interest The authors declare no conflict of interest, financial or otherwise.
Figures
References
-
- Agianniotis A, Stergiopulos N (2012) Wall properties of the apolipoprotein E-deficient mouse aorta. Atherosclerosis 223(2):314–320 - PubMed
-
- Asanuma K, Magid R, Johnson C, Nerem RM, Galis ZS (2003) Uniaxial strain upregulates matrix-degrading enzymes produced by human vascular smooth muscle cells. American Journal of Physiology-Heart and Circulatory Physiology 284(5):H1778–H1784 - PubMed
-
- Bardy N, Merval R, Benessiano J, Samuel JL, Tedgui A (1996) Pressure and angiotensin II synergistically induce aortic fibronectin expression in organ culture model of rabbit aorta: evidence for a pressure-induced tissue renin-angiotensin system. Circulation Research 79(1):70–78 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources