

Short-Chain Fatty Acids Ameliorate Diabetic Nephropathy via GPR43-Mediated Inhibition of Oxidative Stress and NF- κ B Signaling
- PMID: 32831998
- PMCID: PMC7422068
- DOI: 10.1155/2020/4074832
Short-Chain Fatty Acids Ameliorate Diabetic Nephropathy via GPR43-Mediated Inhibition of Oxidative Stress and NF- κ B Signaling
Abstract
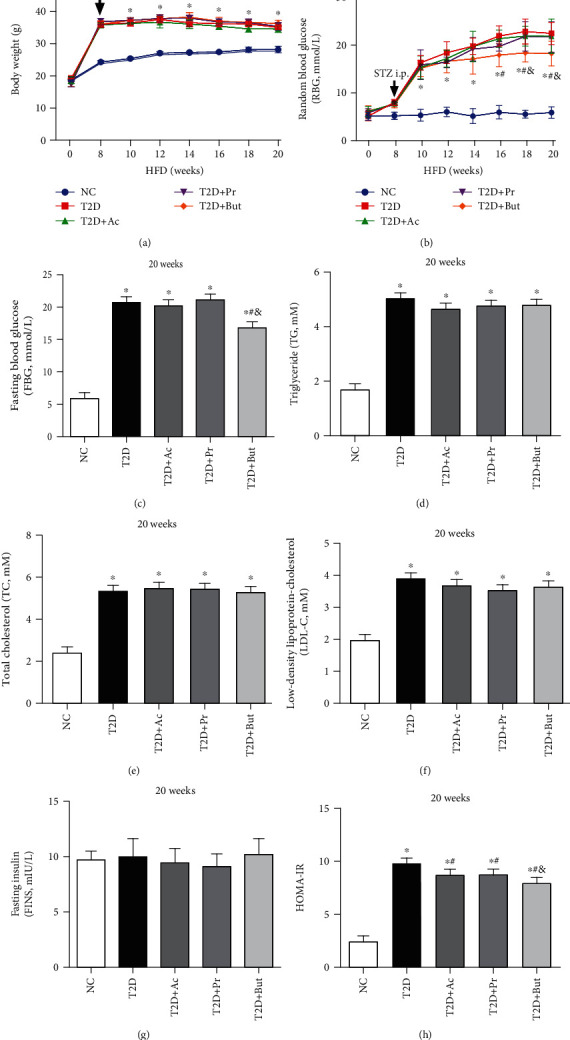
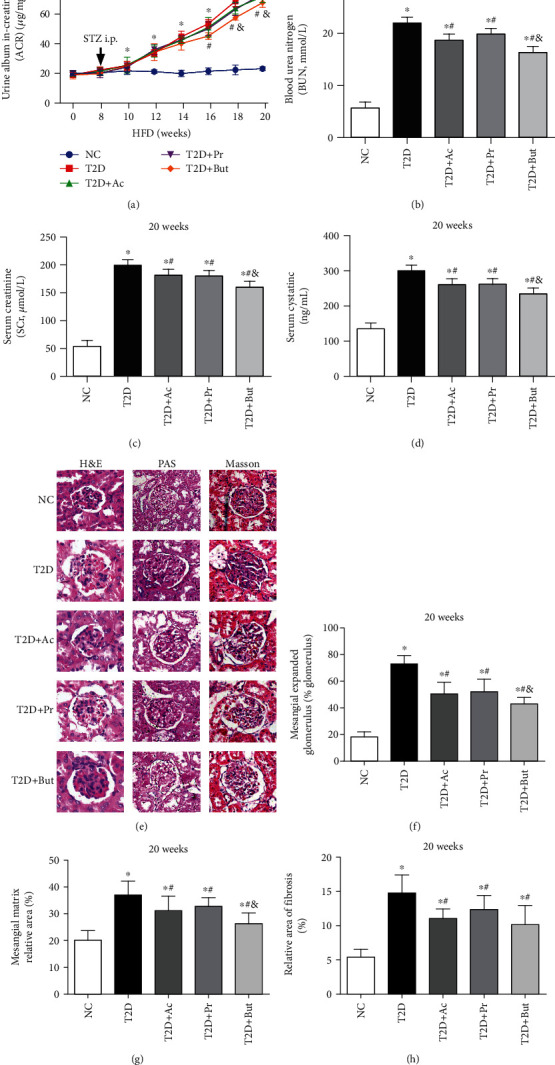
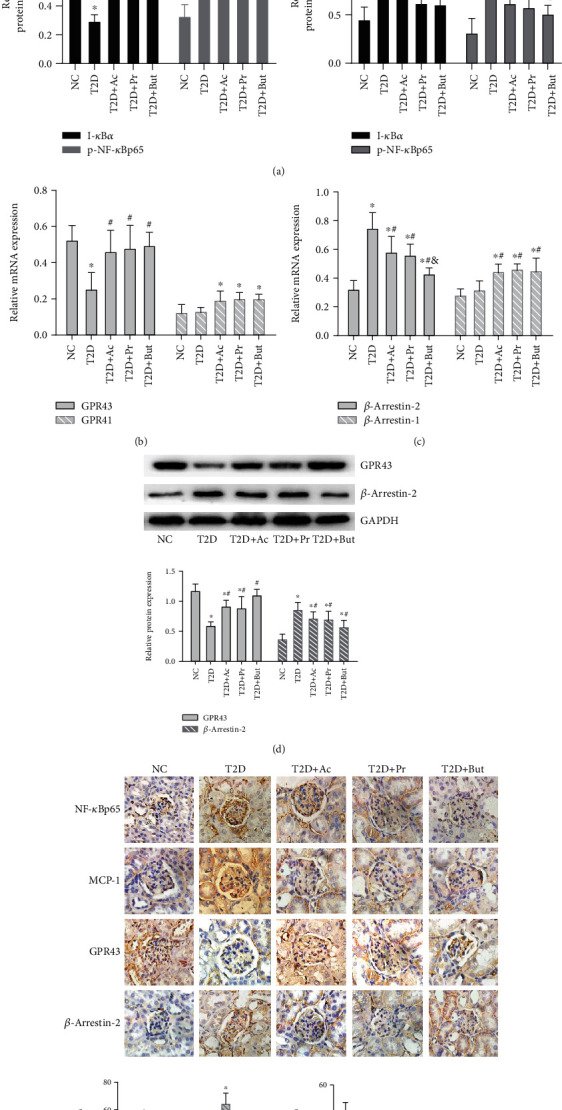
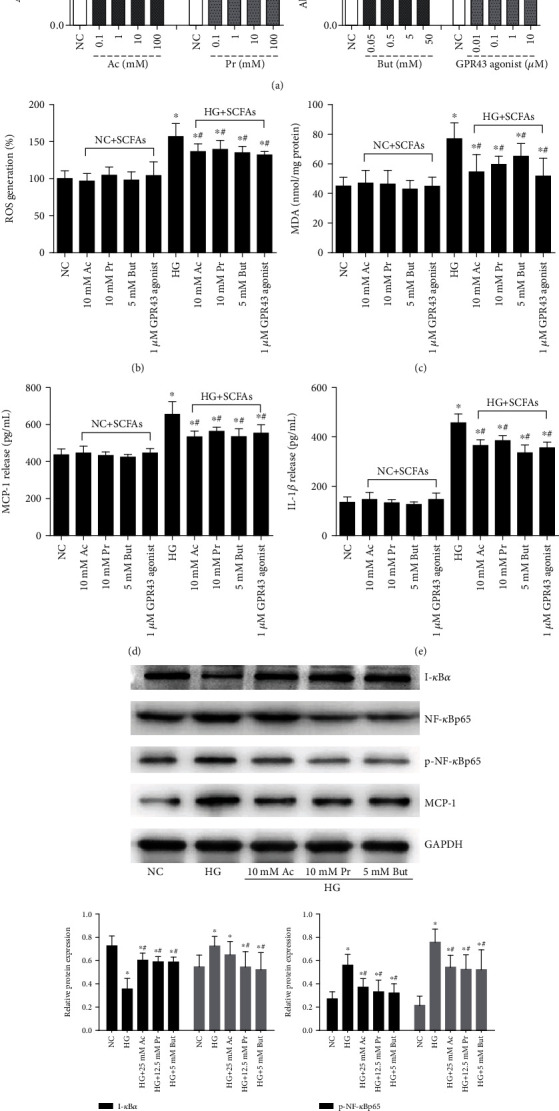
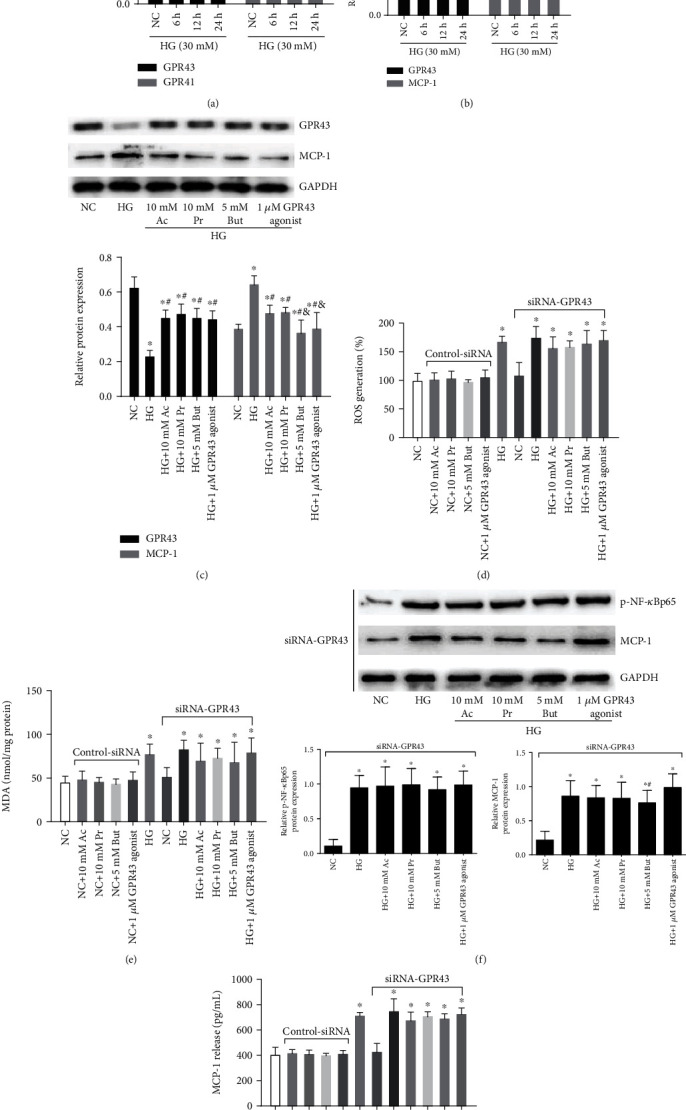
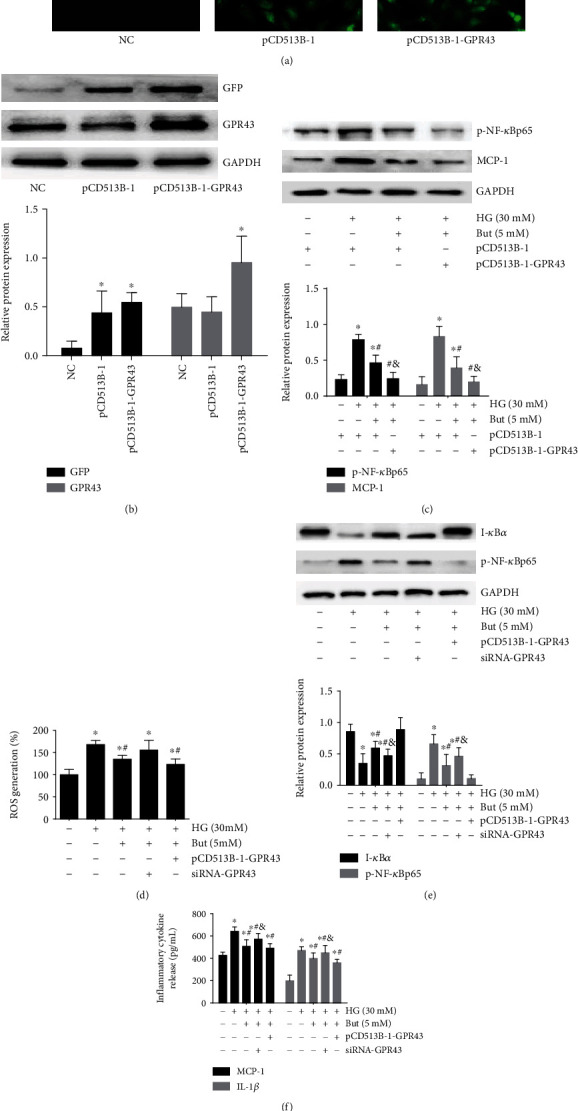
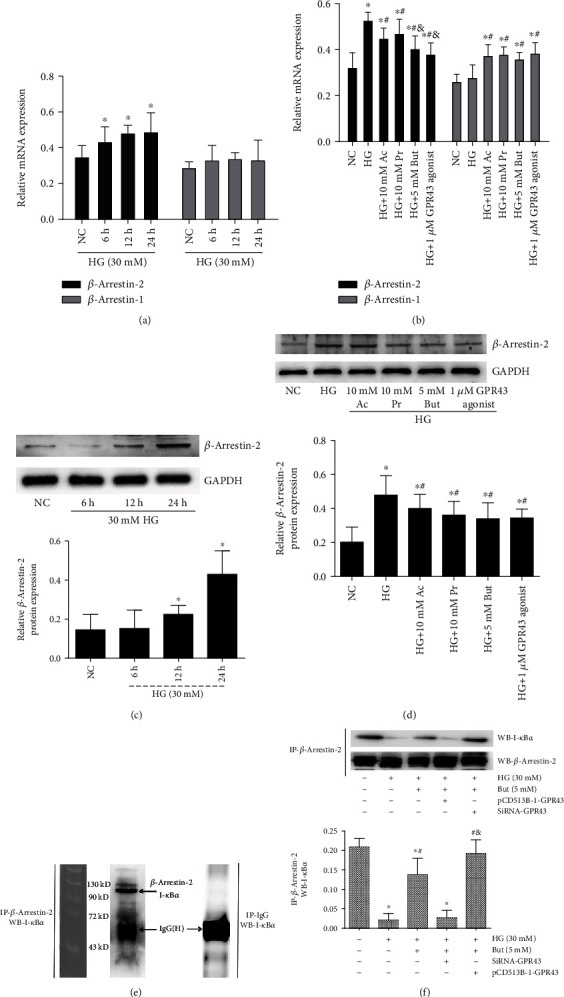
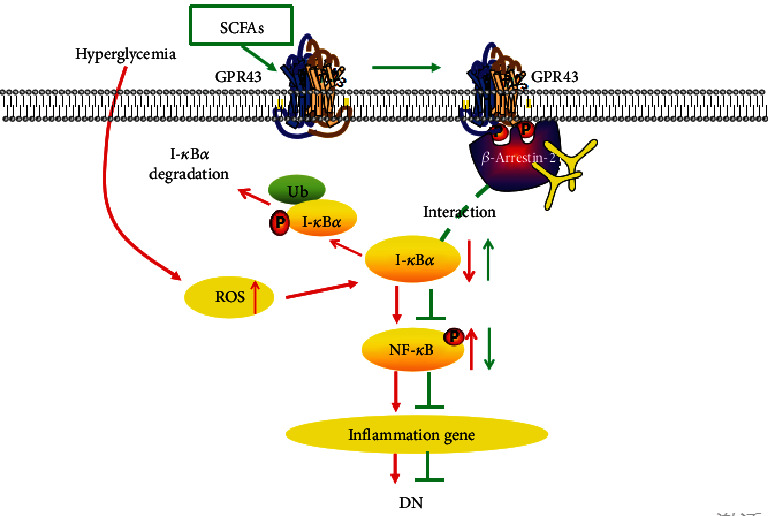
Diabetic nephropathy (DN) is a chronic low-grade inflammatory disease. Oxidative stress and nuclear factor kappa B (NF-κB) signaling play an important role in the pathogenesis of DN. Short-chain fatty acids (SCFAs) produced from carbohydrate fermentation in the gastrointestinal tract exert positive regulatory effects on inflammation and kidney injuries. However, it is unclear whether SCFAs can prevent and ameliorate DN. In the present study, we evaluated the role and mechanism of the three main SCFAs (acetate, propionate, and butyrate) in high-fat diet (HFD) and streptozotocin- (STZ-) induced type2 diabetes (T2D) and DN mouse models and in high glucose-induced mouse glomerular mesangial cells (GMCs), to explore novel therapeutic strategies and molecular targets for DN. We found that exogenous SCFAs, especially butyrate, improved hyperglycemia and insulin resistance; prevented the formation of proteinuria and an increase in serum creatinine, urea nitrogen, and cystatin C; inhibited mesangial matrix accumulation and renal fibrosis; and blocked NF-κB activation in mice. SCFAs also inhibited high glucose-induced oxidative stress and NF-κB activation and enhanced the interaction between β-arrestin-2 and I-κBα in GMCs. Specifically, the beneficial effects of SCFAs were significantly facilitated by the overexpression GPR43 or imitated by a GPR43 agonist but were inhibited by siRNA-GPR43 in GMCs. These results support the conclusion that SCFAs, especially butyrate, partially improve T2D-induced kidney injury via GPR43-mediated inhibition of oxidative stress and NF-κB signaling, suggesting SCFAs may be potential therapeutic agents in the prevention and treatment of DN.
Copyright © 2020 Wei Huang et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
