

Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis
- PMID: 32832598
- PMCID: PMC7439444
- DOI: 10.1126/sciadv.aba1972
Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis
Abstract
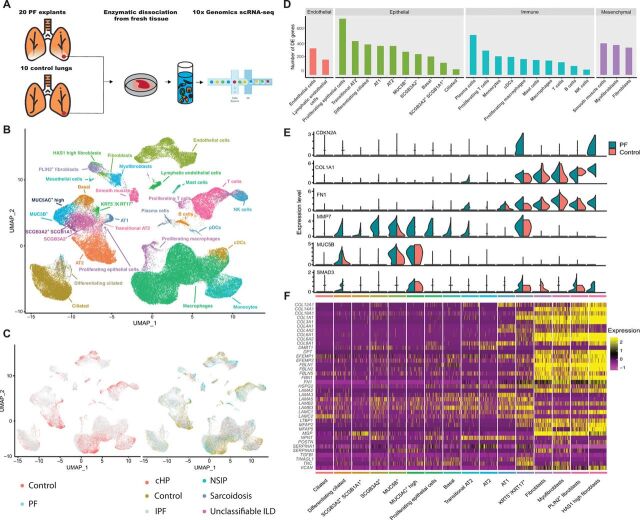
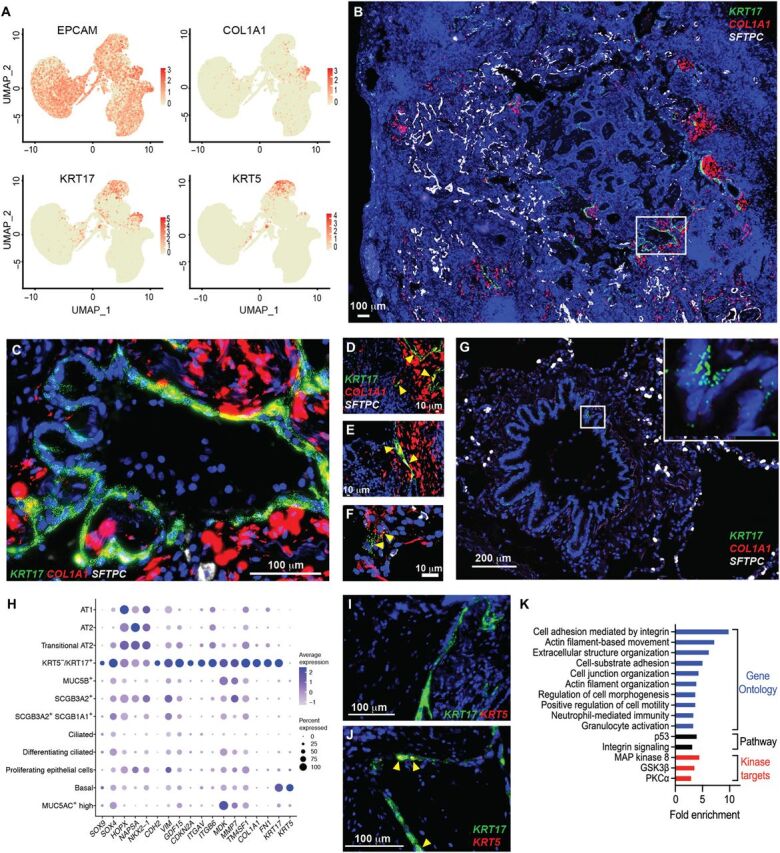
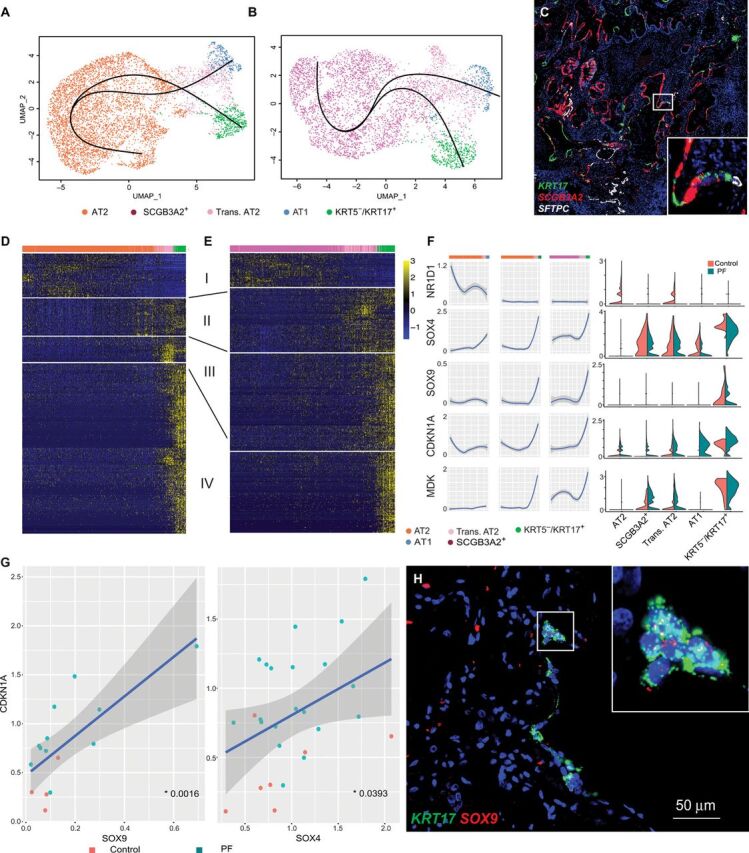
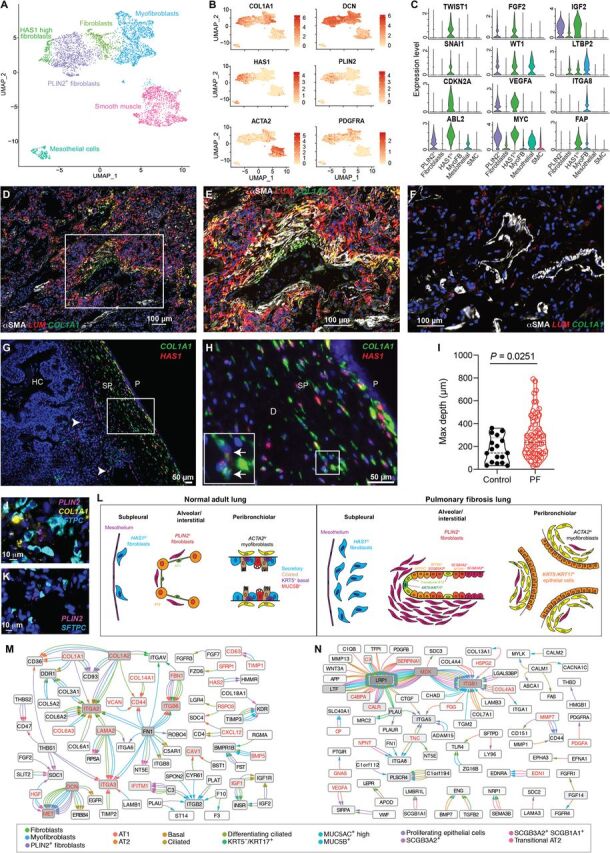
Pulmonary fibrosis (PF) is a form of chronic lung disease characterized by pathologic epithelial remodeling and accumulation of extracellular matrix (ECM). To comprehensively define the cell types, mechanisms, and mediators driving fibrotic remodeling in lungs with PF, we performed single-cell RNA sequencing of single-cell suspensions from 10 nonfibrotic control and 20 PF lungs. Analysis of 114,396 cells identified 31 distinct cell subsets/states. We report that a remarkable shift in epithelial cell phenotypes occurs in the peripheral lung in PF and identify several previously unrecognized epithelial cell phenotypes, including a KRT5- /KRT17 + pathologic, ECM-producing epithelial cell population that was highly enriched in PF lungs. Multiple fibroblast subtypes were observed to contribute to ECM expansion in a spatially discrete manner. Together, these data provide high-resolution insights into the complexity and plasticity of the distal lung epithelium in human disease and indicate a diversity of epithelial and mesenchymal cells contribute to pathologic lung fibrosis.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures


References
-
- Lederer D. J., Martinez F. J., Idiopathic pulmonary fibrosis. N. Engl. J. Med. 378, 1811–1823 (2018). - PubMed
-
- Travis W. D., Costabel U., Hansell D. M., King Jr. T. E., Lynch D. A., Nicholson A. G., Ryerson C. J., Ryu J. H., Selman M., Wells A. U., Behr J., Bouros D., Brown K. K., Colby T. V., Collard H. R., Cordeiro C. R., Cottin V., Crestani B., Drent M., Dudden R. F., Egan J., Flaherty K., Hogaboam C., Inoue Y., Johkoh T., Kim D. S., Kitaichi M., Loyd J., Martinez F. J., Myers J., Protzko S., Raghu G., Richeldi L., Sverzellati N., Swigris J., Valeyre D.; ATS/ERS Committee on Idiopathic Interstitial Pneumonias , An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 188, 733–748 (2013). - PMC - PubMed
-
- Thomas A. Q., Lane K., Phillips J. III, Prince M., Markin C., Speer M., Schwartz D. A., Gaddipati R., Marney A., Johnson J., Roberts R., Haines J., Stahlman M., Loyd J. E., Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 165, 1322–1328 (2002). - PubMed
Publication types
MeSH terms
Grants and funding
- K08 HL130595/HL/NHLBI NIH HHS/United States
- R01 HL126176/HL/NHLBI NIH HHS/United States
- P30 DK058404/DK/NIDDK NIH HHS/United States
- R01 HL145372/HL/NHLBI NIH HHS/United States
- K08 HL136888/HL/NHLBI NIH HHS/United States
- K08 HL143051/HL/NHLBI NIH HHS/United States
- P30 DK020593/DK/NIDDK NIH HHS/United States
- R01 HL151016/HL/NHLBI NIH HHS/United States
- IK2 BX003841/BX/BLRD VA/United States
- I01 BX002378/BX/BLRD VA/United States
- U24 DK059637/DK/NIDDK NIH HHS/United States
- P01 HL092870/HL/NHLBI NIH HHS/United States
- UL1 TR000445/TR/NCATS NIH HHS/United States
- P30 EY008126/EY/NEI NIH HHS/United States
- T32 HL094296/HL/NHLBI NIH HHS/United States
- P30 CA068485/CA/NCI NIH HHS/United States
- P30 AI110527/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
