

An information gain-based approach for evaluating protein structure models
- PMID: 32837711
- PMCID: PMC7431362
- DOI: 10.1016/j.csbj.2020.08.013
An information gain-based approach for evaluating protein structure models
Abstract
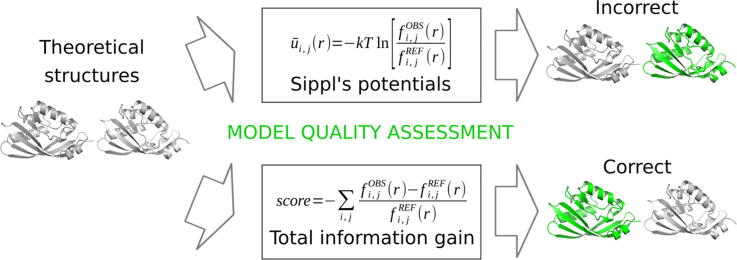
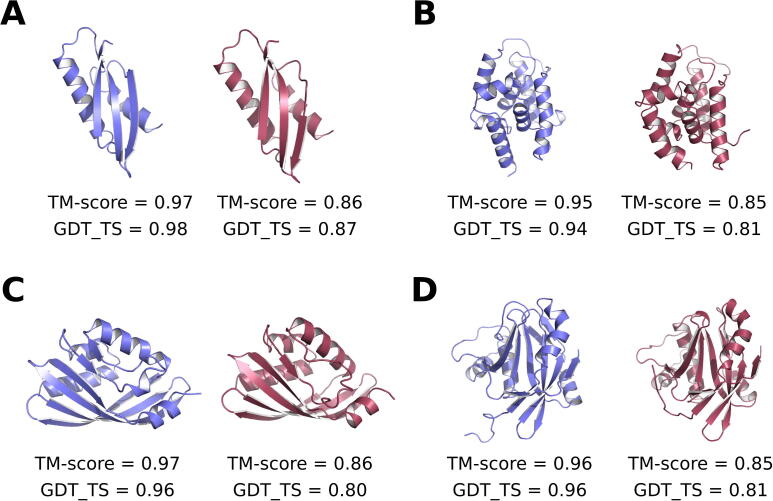
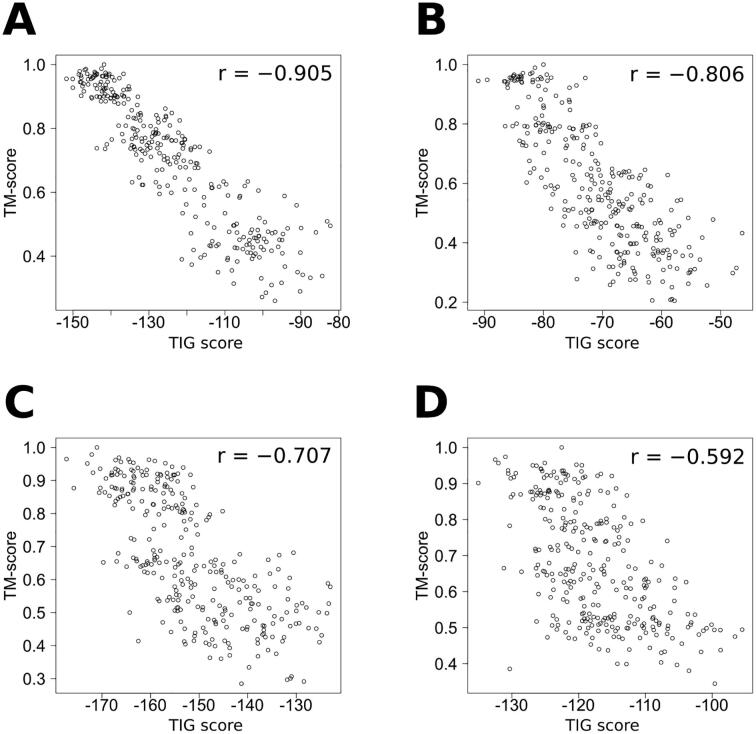
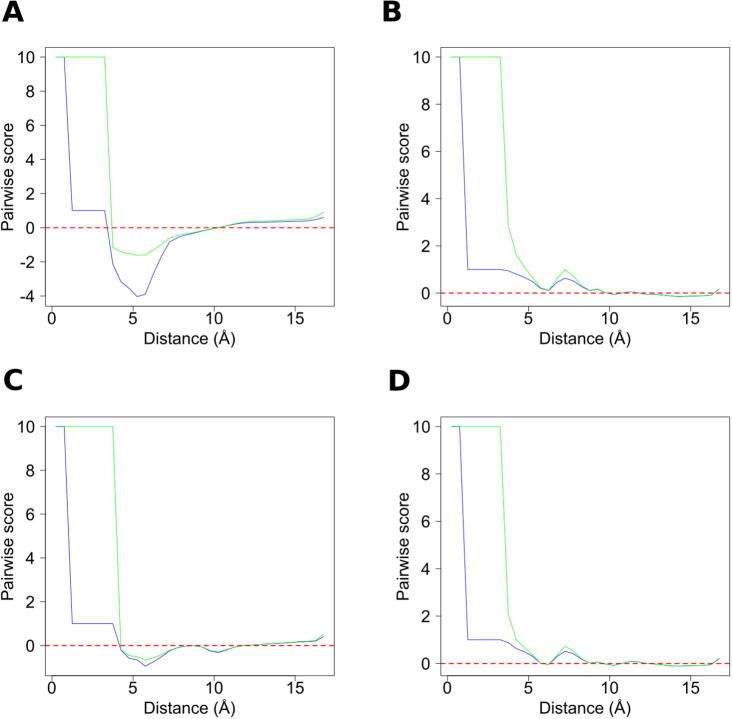
For three decades now, knowledge-based scoring functions that operate through the "potential of mean force" (PMF) approach have continuously proven useful for studying protein structures. Although these statistical potentials are not to be confused with their physics-based counterparts of the same name-i.e. PMFs obtained by molecular dynamics simulations-their particular success in assessing the native-like character of protein structure predictions has lead authors to consider the computed scores as approximations of the free energy. However, this physical justification is a matter of controversy since the beginning. Alternative interpretations based on Bayes' theorem have been proposed, but the misleading formalism that invokes the inverse Boltzmann law remains recurrent in the literature. In this article, we present a conceptually new method for ranking protein structure models by quality, which is (i) independent of any physics-based explanation and (ii) relevant to statistics and to a general definition of information gain. The theoretical development described in this study provides new insights into how statistical PMFs work, in comparison with our approach. To prove the concept, we have built interatomic distance-dependent scoring functions, based on the former and new equations, and compared their performance on an independent benchmark of 60,000 protein structures. The results demonstrate that our new formalism outperforms statistical PMFs in evaluating the quality of protein structural decoys. Therefore, this original type of score offers a possibility to improve the success of statistical PMFs in the various fields of structural biology where they are applied. The open-source code is available for download at https://gitlab.rpbs.univ-paris-diderot.fr/src/ig-score.
Keywords: Knowledge-based scoring functions; Model quality assessment; Protein structure prediction; Statistical potentials.
© 2020 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
MyPMFs: a simple tool for creating statistical potentials to assess protein structural models.Biochimie. 2018 Aug;151:37-41. doi: 10.1016/j.biochi.2018.05.013. Epub 2018 May 29. Biochimie. 2018. PMID: 29857183
-
Potentials of mean force for protein structure prediction vindicated, formalized and generalized.PLoS One. 2010 Nov 10;5(11):e13714. doi: 10.1371/journal.pone.0013714. PLoS One. 2010. PMID: 21103041 Free PMC article.
-
Are distance-dependent statistical potentials considering three interacting bodies superior to two-body statistical potentials for protein structure prediction?J Bioinform Comput Biol. 2014 Oct;12(5):1450022. doi: 10.1142/S021972001450022X. Epub 2014 Sep 11. J Bioinform Comput Biol. 2014. PMID: 25212727
-
Fast protein fragment similarity scoring using a Binet-Cauchy kernel.Bioinformatics. 2014 Mar 15;30(6):784-91. doi: 10.1093/bioinformatics/btt618. Epub 2013 Oct 27. Bioinformatics. 2014. PMID: 24167157
-
dMM-PBSA: A New HADDOCK Scoring Function for Protein-Peptide Docking.Front Mol Biosci. 2016 Aug 31;3:46. doi: 10.3389/fmolb.2016.00046. eCollection 2016. Front Mol Biosci. 2016. PMID: 27630991 Free PMC article.
Cited by
-
An integrated protein structure fitness scoring approach for identifying native-like model structures.Comput Struct Biotechnol J. 2022 Nov 17;20:6467-6472. doi: 10.1016/j.csbj.2022.11.032. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36467582 Free PMC article.
-
Representations of protein structure for exploring the conformational space: A speed-accuracy trade-off.Comput Struct Biotechnol J. 2021 Apr 28;19:2618-2625. doi: 10.1016/j.csbj.2021.04.049. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 34025948 Free PMC article.
-
Hierarchical Analysis of Protein Structures: From Secondary Structures to Protein Units and Domains.Methods Mol Biol. 2025;2870:357-370. doi: 10.1007/978-1-0716-4213-9_18. Methods Mol Biol. 2025. PMID: 39543044
References
LinkOut - more resources
Full Text Sources
Miscellaneous