

The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut
- PMID: 32841606
- PMCID: PMC7443397
- DOI: 10.1016/j.chom.2020.08.003
The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut
Abstract
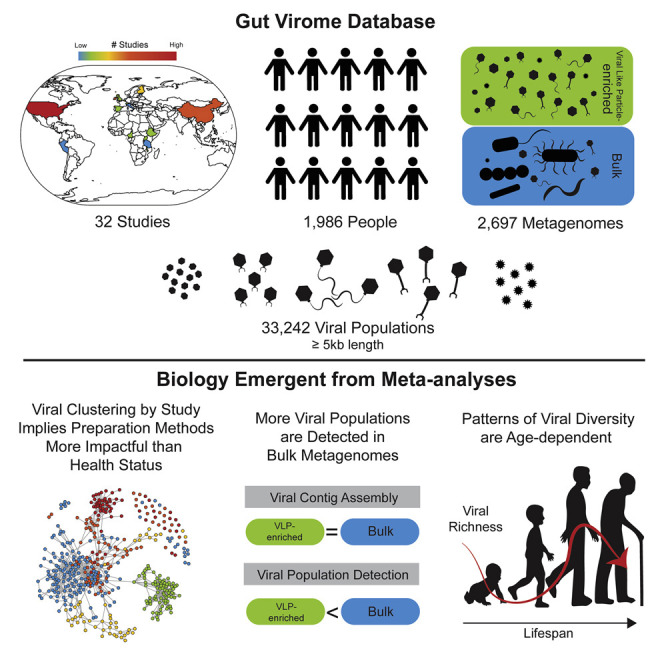
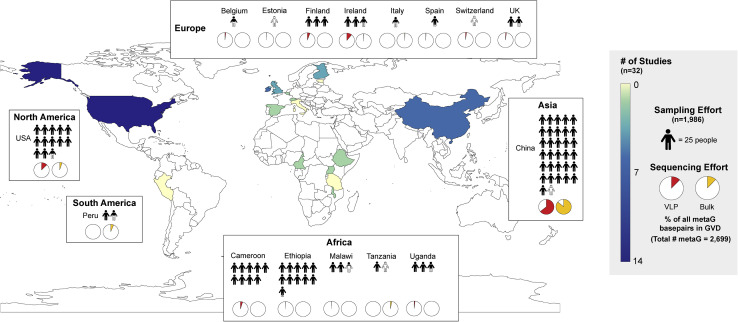
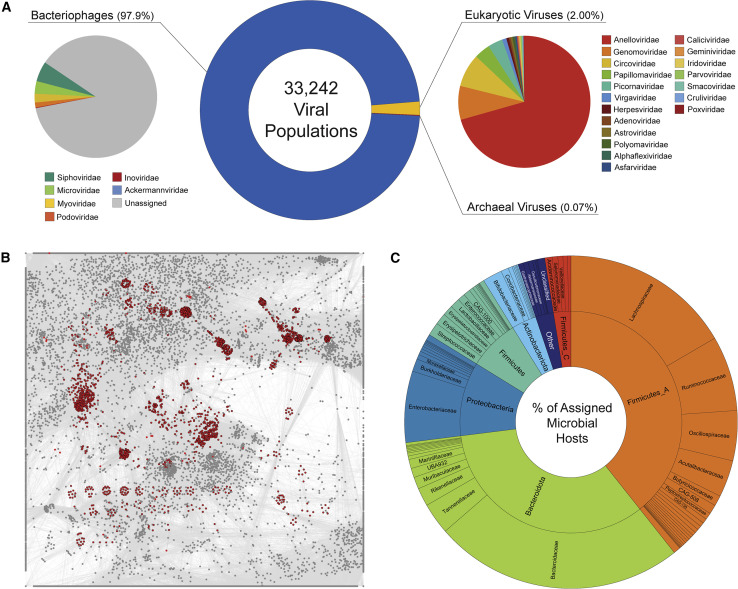
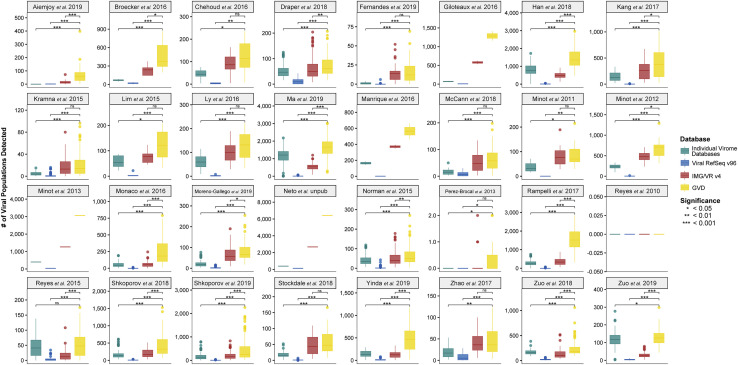
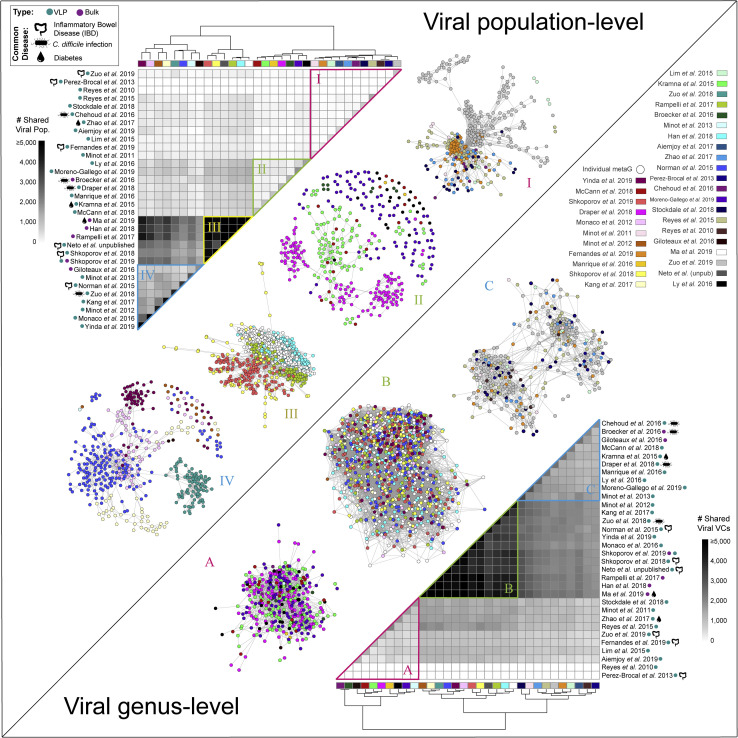
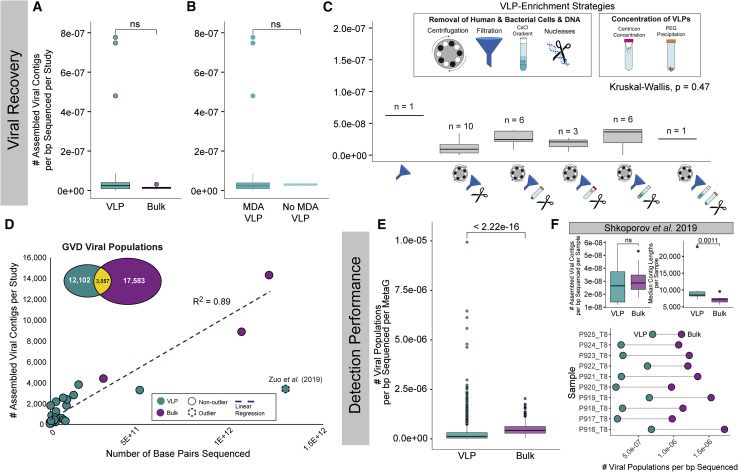
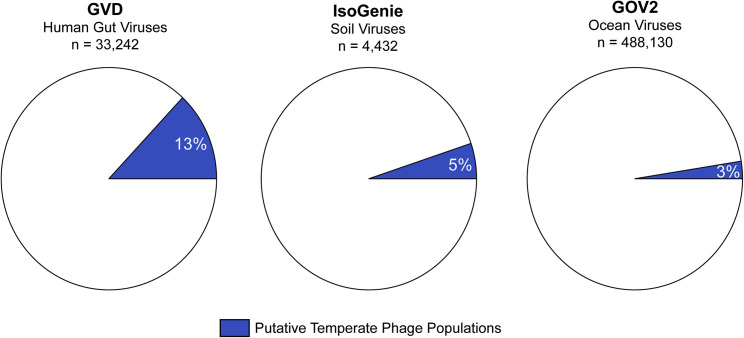
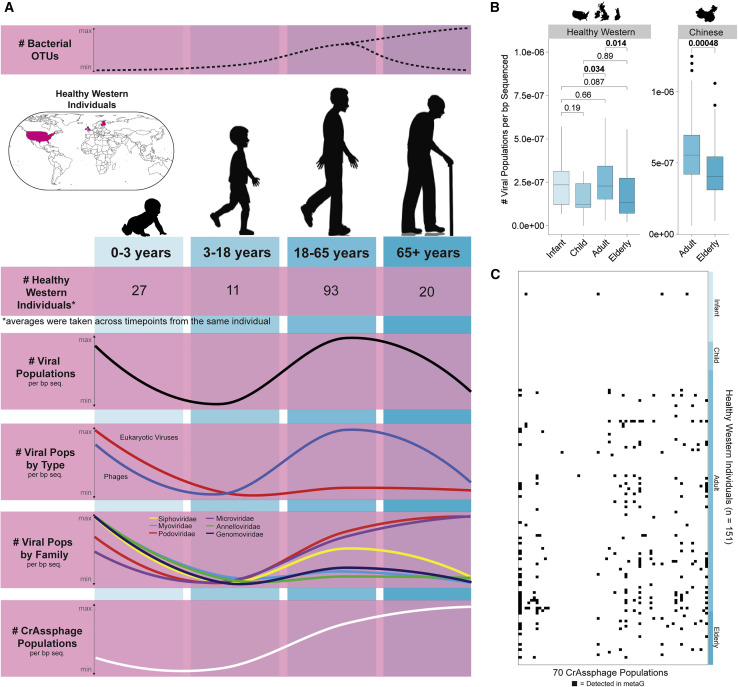
The gut microbiome profoundly affects human health and disease, and their infecting viruses are likely as important, but often missed because of reference database limitations. Here, we (1) built a human Gut Virome Database (GVD) from 2,697 viral particle or microbial metagenomes from 1,986 individuals representing 16 countries, (2) assess its effectiveness, and (3) report a meta-analysis that reveals age-dependent patterns across healthy Westerners. The GVD contains 33,242 unique viral populations (approximately species-level taxa) and improves average viral detection rates over viral RefSeq and IMG/VR nearly 182-fold and 2.6-fold, respectively. GVD meta-analyses show highly personalized viromes, reveal that inter-study variability from technical artifacts is larger than any "disease" effect at the population level, and document how viral diversity changes from human infancy into senescence. Together, this compact foundational resource, these standardization guidelines, and these meta-analysis findings provide a systematic toolkit to help maximize our understanding of viral roles in health and disease.
Keywords: bacteriophage; database; dysbiosis; gut microbiome; human health; lifespan; virome; virus.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Comment in
-
A database to identify the human gut virome.Nat Microbiol. 2023 Jan;8(1):5. doi: 10.1038/s41564-022-01280-z. Nat Microbiol. 2023. PMID: 36550315 No abstract available.
References
-
- Adriaenssens E.M., Sullivan M.B., Knezevic P., Van Zyl L.J. 2020. Taxonomy of prokaryotic viruses: 2018 – 2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
