

A genetic barcode of SARS-CoV-2 for monitoring global distribution of different clades during the COVID-19 pandemic
- PMID: 32841689
- PMCID: PMC7443060
- DOI: 10.1016/j.ijid.2020.08.052
A genetic barcode of SARS-CoV-2 for monitoring global distribution of different clades during the COVID-19 pandemic
Abstract
Objective: The SARS-CoV-2 pathogen has established endemicity in humans. This necessitates the development of rapid genetic surveillance methodologies to serve as an adjunct with existing comprehensive, albeit though slower, genome sequencing-driven approaches.
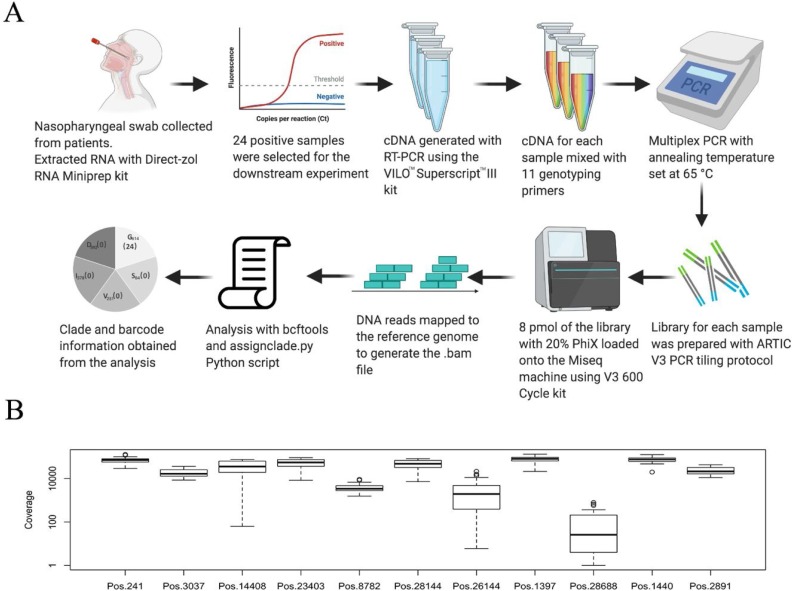
Methods: A total of 21,789 complete genomes were downloaded from GISAID on May 28, 2020 for analyses. We have defined the major clades and subclades of circulating SARS-CoV-2 genomes. A rapid sequencing-based genotyping protocol was developed and tested on SARS-CoV-2-positive RNA samples by next-generation sequencing.
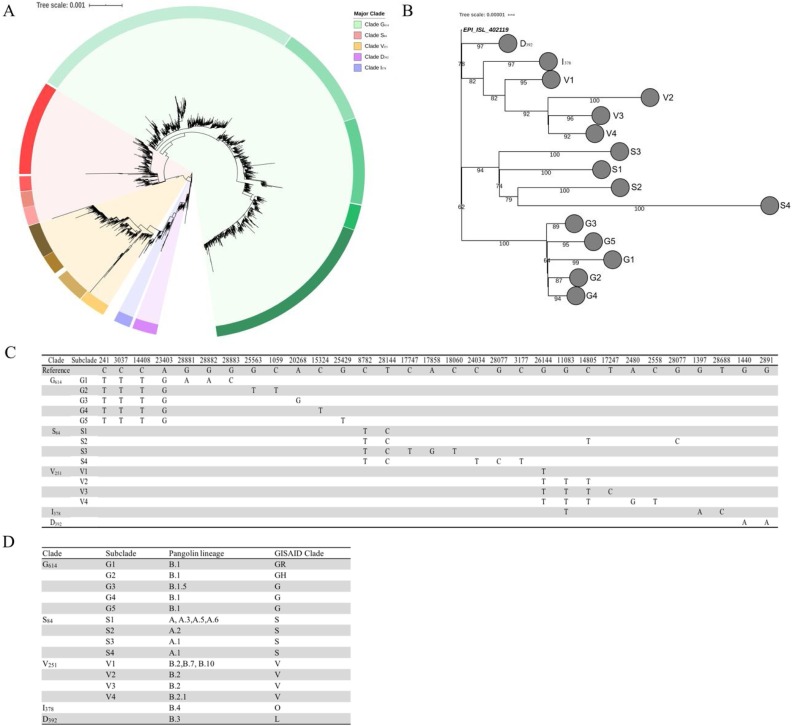
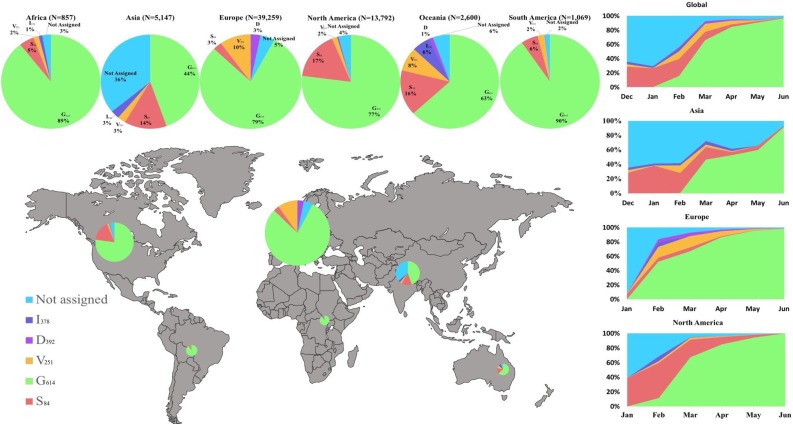
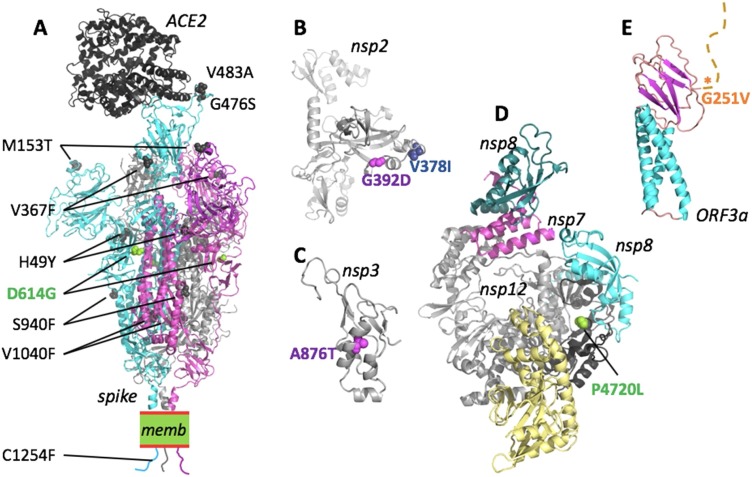
Results: We describe 11 major mutations which defined five major clades (G614, S84, V251, I378 and D392) of globally circulating viral populations. The clades can specifically identify using an 11-nucleotide genetic barcode. An analysis of amino acid variation in SARS-CoV-2 proteins provided evidence of substitution events in the viral proteins involved in both host entry and genome replication.
Conclusion: Globally circulating SARS-CoV-2 genomes could be classified into 5 major clades based on mutational profiles defined by an 11-nucleotide barcode. We have successfully developed a multiplexed sequencing-based, rapid genotyping protocol for high-throughput classification of major clade types of SARS-CoV-2 in clinical samples. This barcoding strategy will be required to monitor decreases in genetic diversity as treatment and vaccine approaches become widely available.
Keywords: SARS-CoV-2; barcoding; genetic surveillance; genome variation.
Copyright © 2020 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
References
-
- Arnold K., Bordoli L., Kopp J., Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
