

Inferring clonal composition from multiple tumor biopsies
- PMID: 32843649
- PMCID: PMC7447821
- DOI: 10.1038/s41540-020-00147-5
Inferring clonal composition from multiple tumor biopsies
Abstract
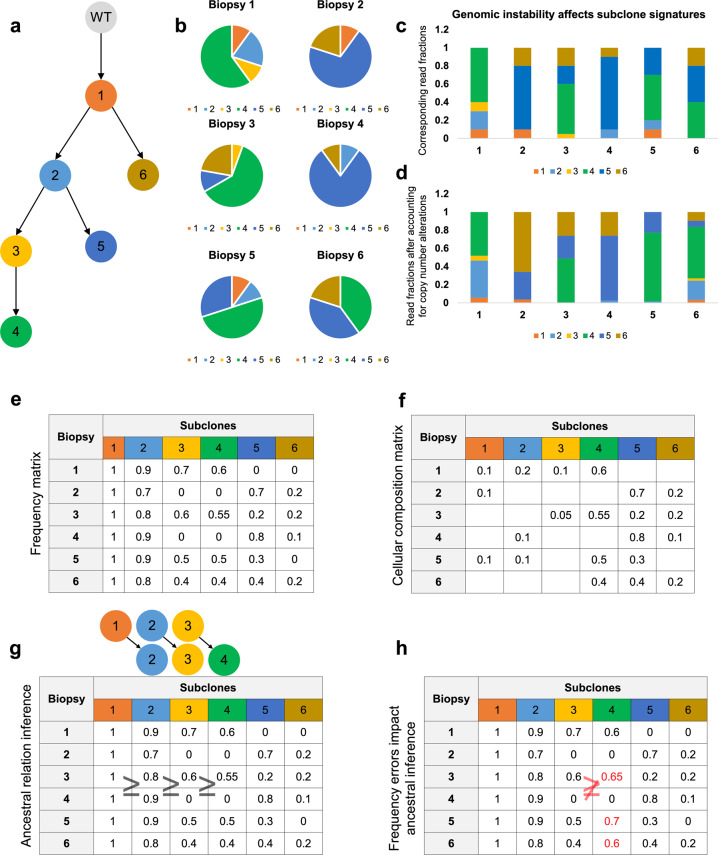
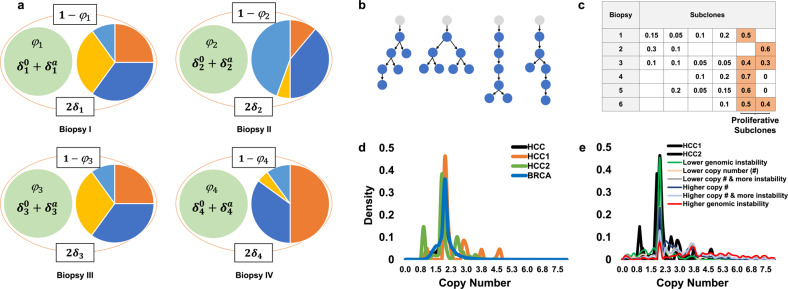
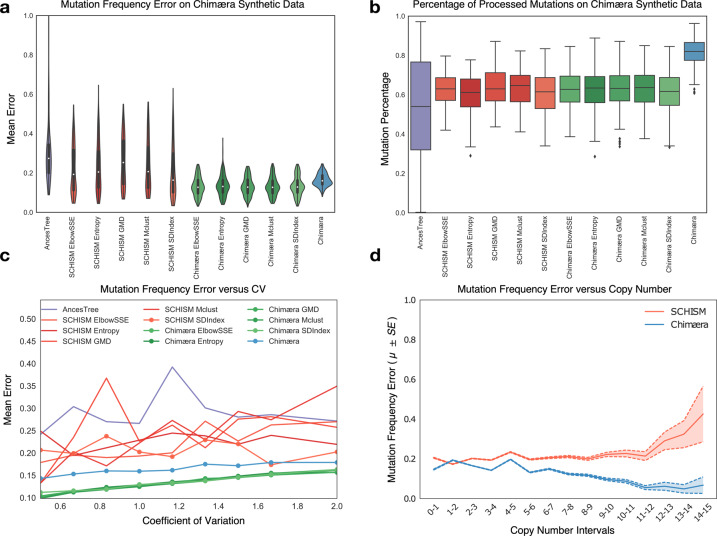
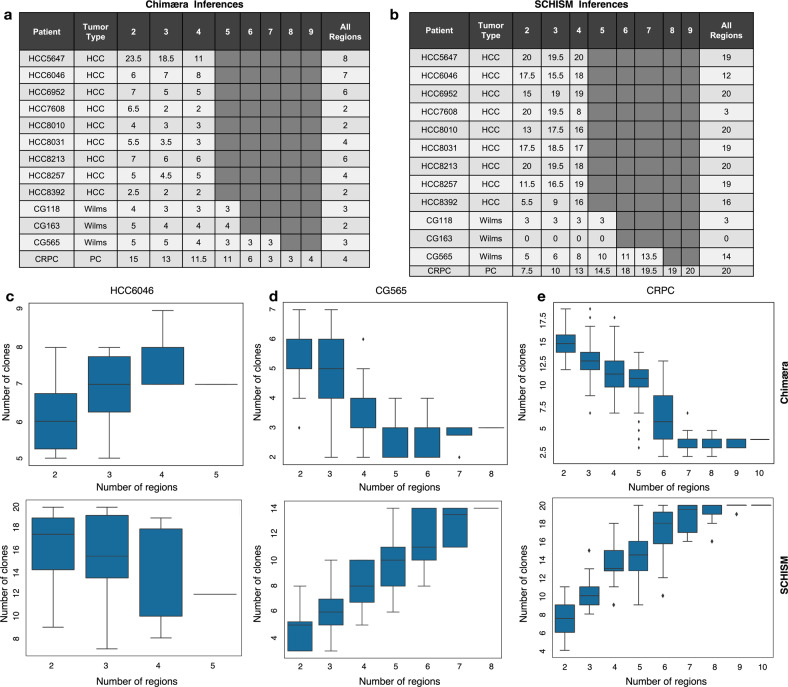
Knowledge about the clonal evolution of a tumor can help to interpret the function of its genetic alterations by identifying initiating events and events that contribute to the selective advantage of proliferative, metastatic, and drug-resistant subclones. Clonal evolution can be reconstructed from estimates of the relative abundance (frequency) of subclone-specific alterations in tumor biopsies, which, in turn, inform on its composition. However, estimating these frequencies is complicated by the high genetic instability that characterizes many cancers. Models for genetic instability suggest that copy number alterations (CNAs) can influence mutation-frequency estimates and thus impede efforts to reconstruct tumor phylogenies. Our analysis suggested that accurate mutation frequency estimates require accounting for CNAs-a challenging endeavour using the genetic profile of a single tumor biopsy. Instead, we propose an optimization algorithm, Chimæra, to account for the effects of CNAs using profiles of multiple biopsies per tumor. Analyses of simulated data and tumor profiles suggested that Chimæra estimates are consistently more accurate than those of previously proposed methods and resulted in improved phylogeny reconstructions and subclone characterizations. Our analyses inferred recurrent initiating mutations in hepatocellular carcinomas, resolved the clonal composition of Wilms' tumors, and characterized the acquisition of mutations in drug-resistant prostate cancers.
Conflict of interest statement
The authors declare no competing interests.
Figures
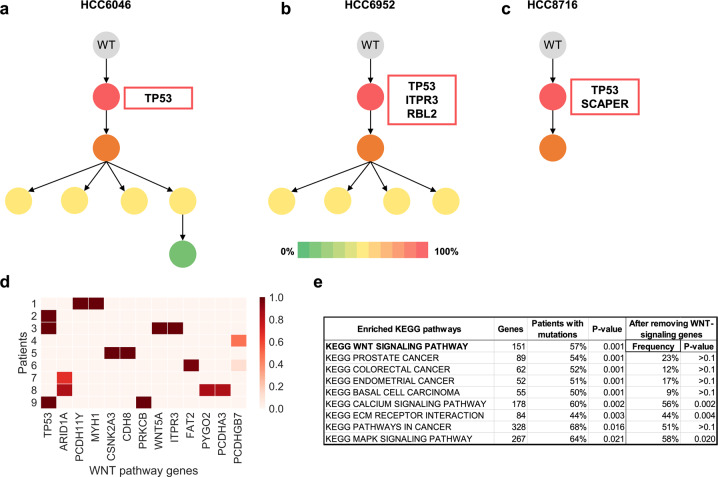
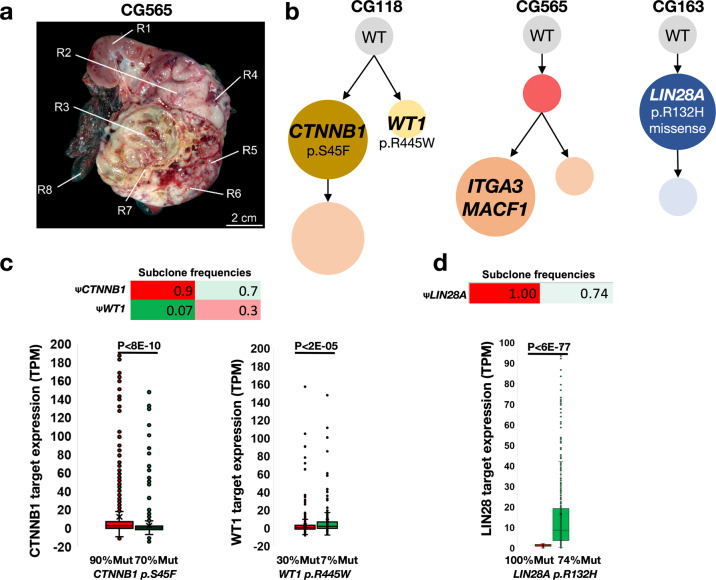
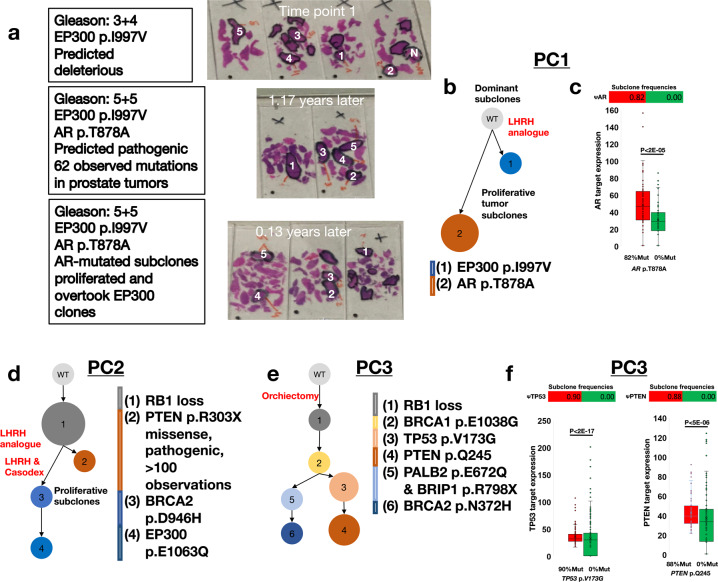
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
