

A loss-of-function NUAK2 mutation in humans causes anencephaly due to impaired Hippo-YAP signaling
- PMID: 32845958
- PMCID: PMC7953732
- DOI: 10.1084/jem.20191561
A loss-of-function NUAK2 mutation in humans causes anencephaly due to impaired Hippo-YAP signaling
Abstract
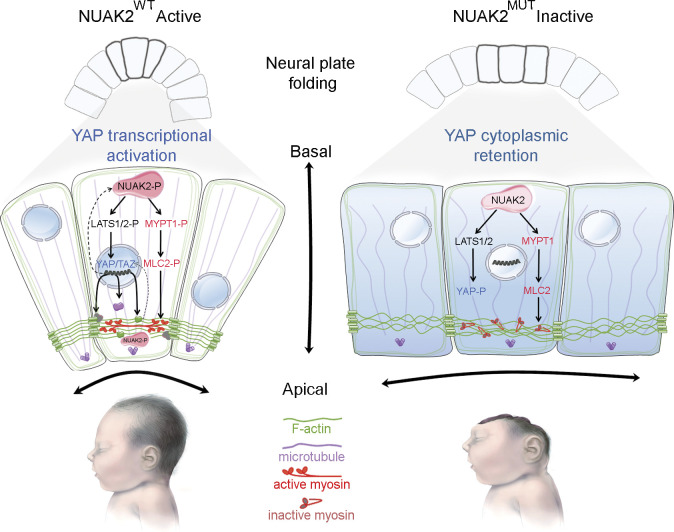
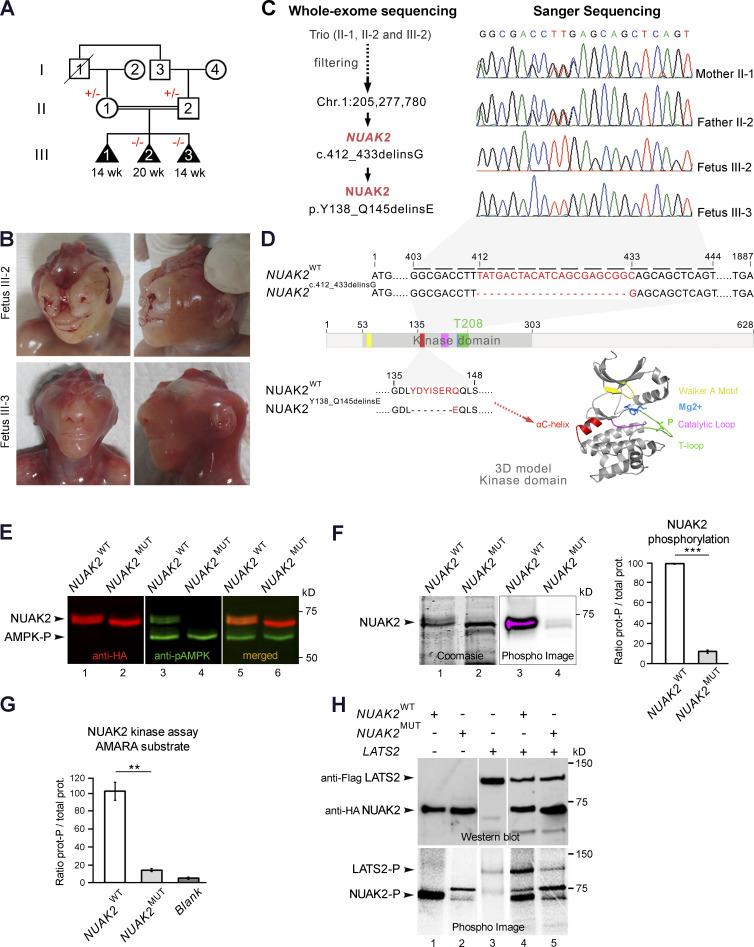
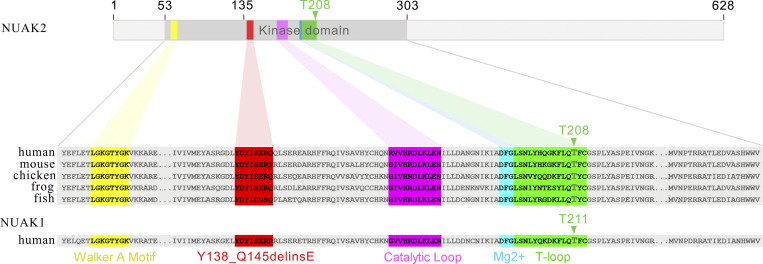
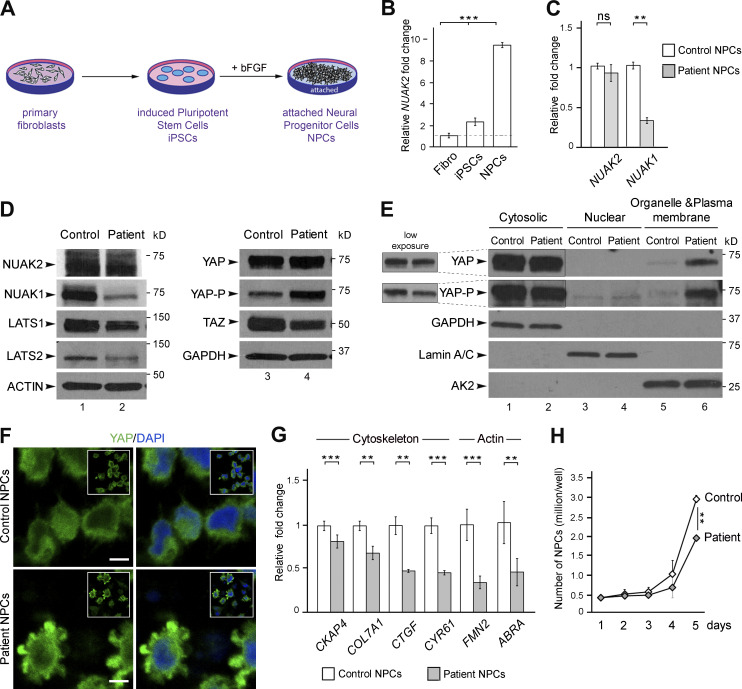
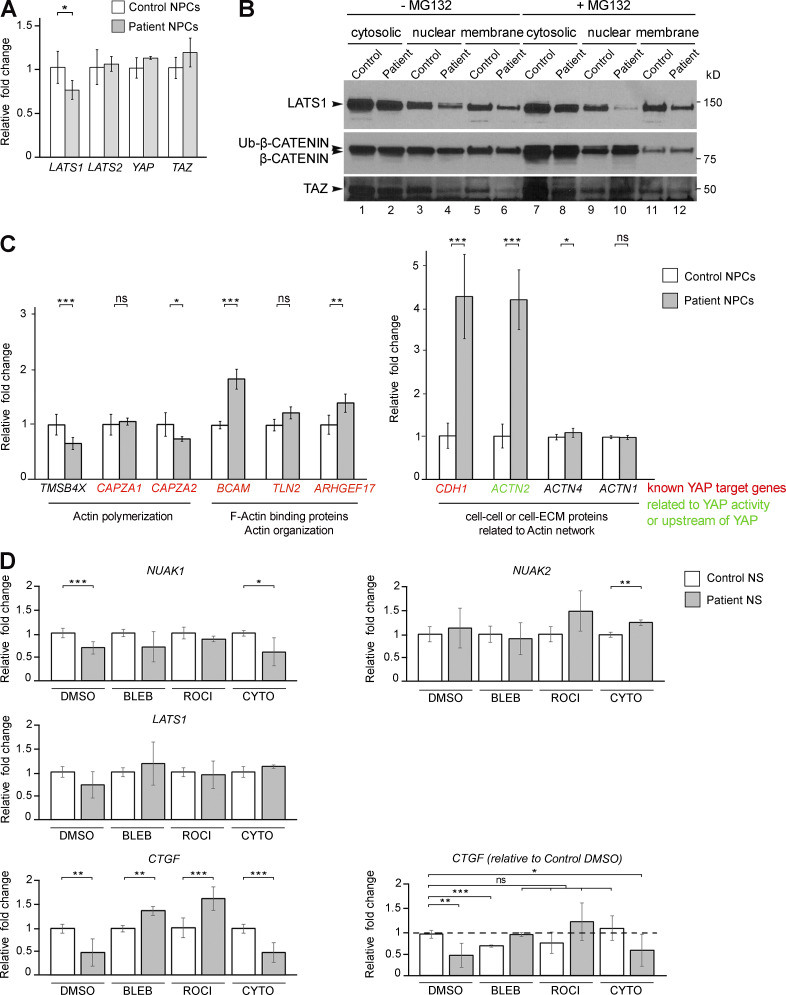
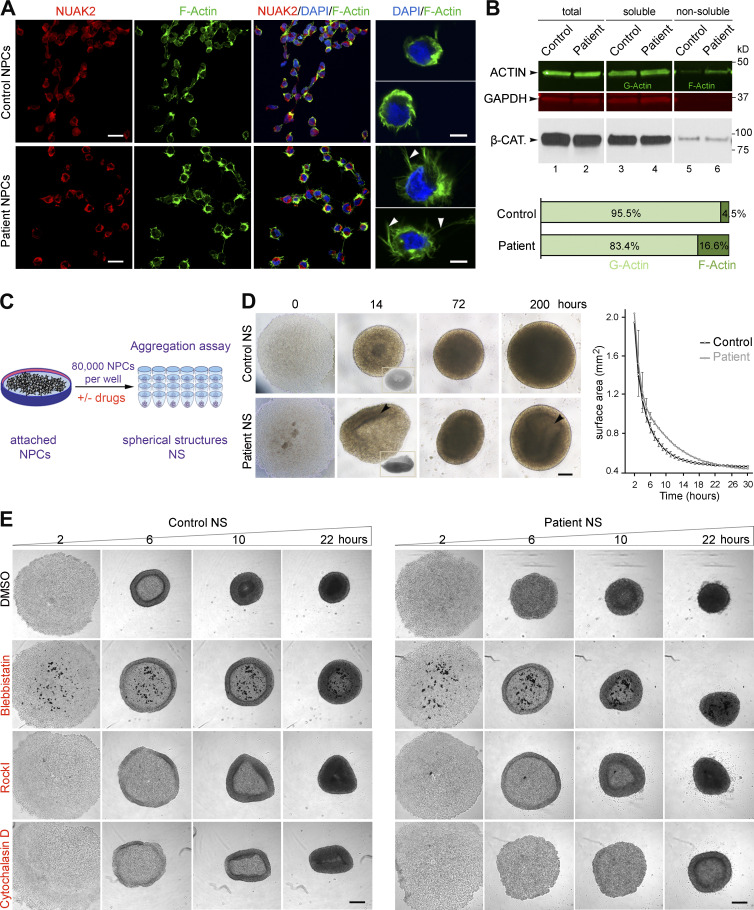
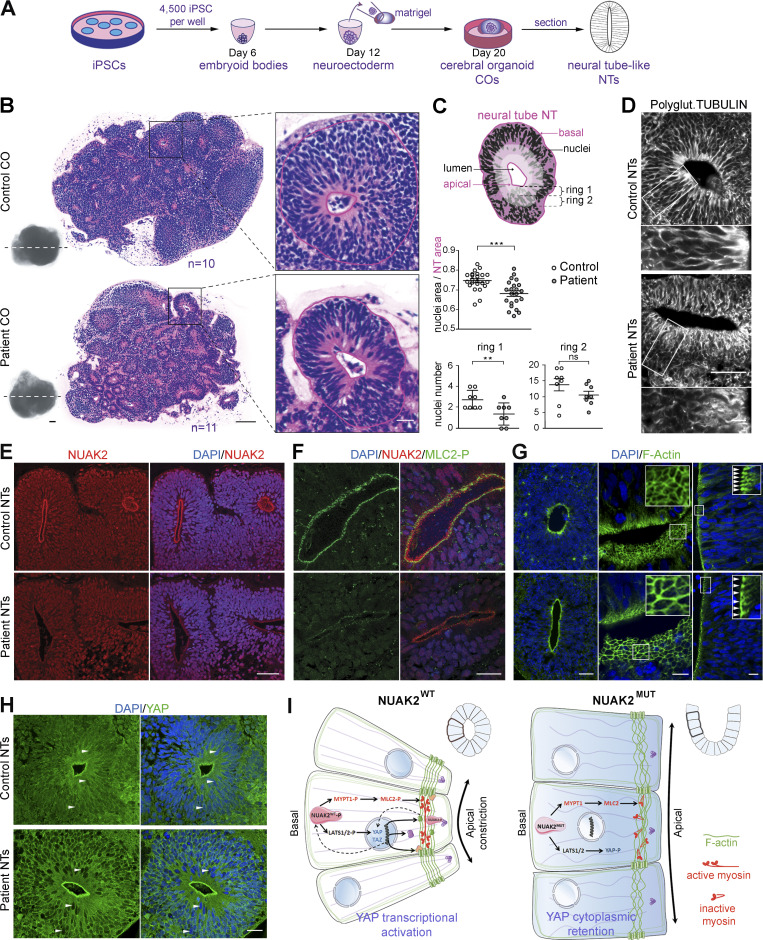
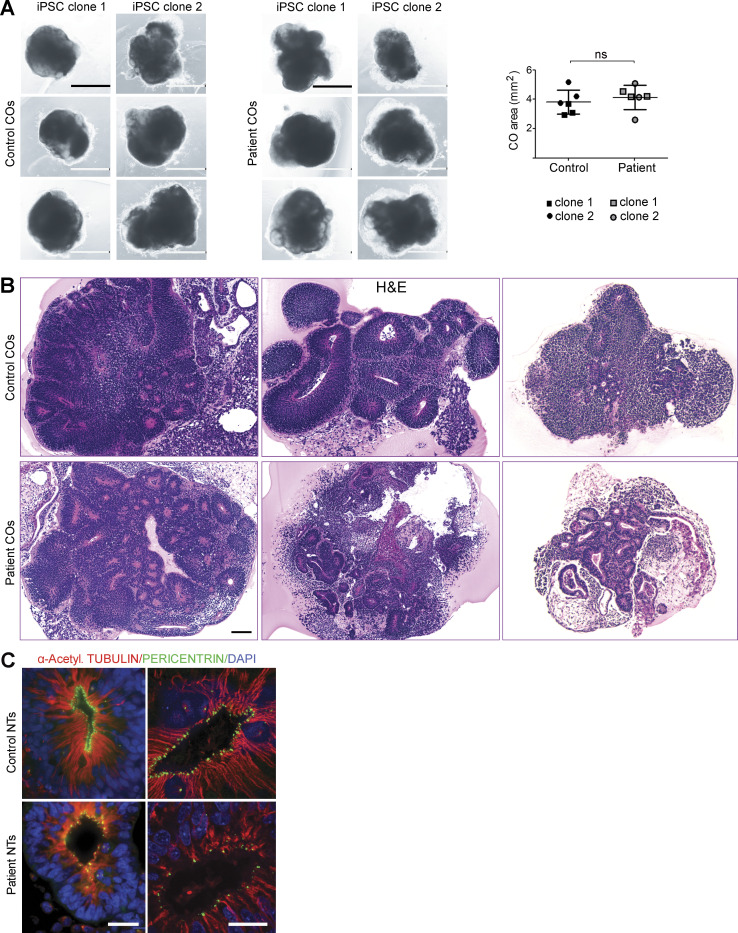
Failure of neural tube closure during embryonic development can result in anencephaly, one of the most common birth defects in humans. A family with recurrent anencephalic fetuses was investigated to understand its etiology and pathogenesis. Exome sequencing revealed a recessive germline 21-bp in-frame deletion in NUAK2 segregating with the disease. In vitro kinase assays demonstrated that the 7-amino acid truncation in NUAK2, a serine/threonine kinase, completely abrogated its catalytic activity. Patient-derived disease models including neural progenitor cells and cerebral organoids showed that loss of NUAK2 activity led to decreased Hippo signaling via cytoplasmic YAP retention. In neural tube-like structures, endogenous NUAK2 colocalized apically with the actomyosin network, which was disrupted in patient cells, causing impaired nucleokinesis and apical constriction. Our results establish NUAK2 as an indispensable kinase for brain development in humans and suggest that a NUAK2-Hippo signaling axis regulates cytoskeletal processes that govern cell shape during neural tube closure.
© 2020 Bonnard et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
