

CFTR trafficking mutations disrupt cotranslational protein folding by targeting biosynthetic intermediates
- PMID: 32848127
- PMCID: PMC7450043
- DOI: 10.1038/s41467-020-18101-8
CFTR trafficking mutations disrupt cotranslational protein folding by targeting biosynthetic intermediates
Abstract
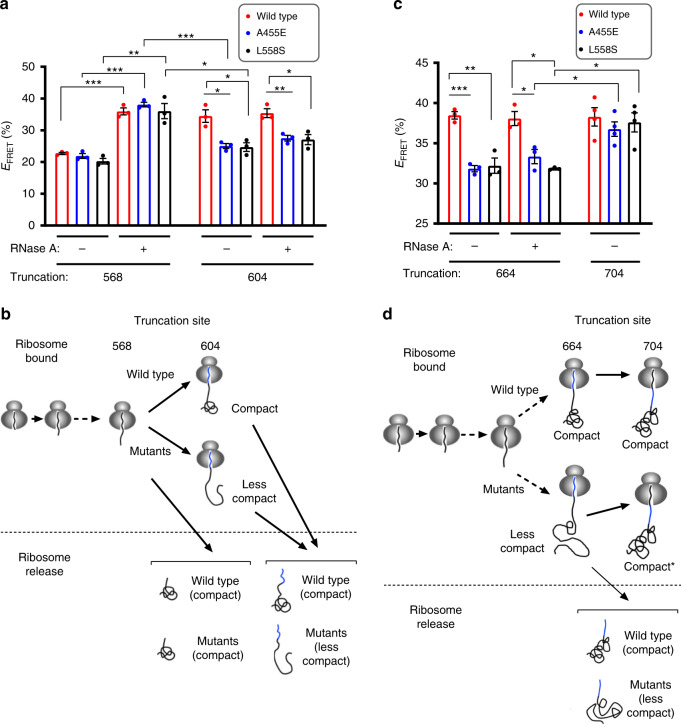
Protein misfolding causes a wide spectrum of human disease, and therapies that target misfolding are transforming the clinical care of cystic fibrosis. Despite this success, however, very little is known about how disease-causing mutations affect the de novo folding landscape. Here we show that inherited, disease-causing mutations located within the first nucleotide-binding domain (NBD1) of the cystic fibrosis transmembrane conductance regulator (CFTR) have distinct effects on nascent polypeptides. Two of these mutations (A455E and L558S) delay compaction of the nascent NBD1 during a critical window of synthesis. The observed folding defect is highly dependent on nascent chain length as well as its attachment to the ribosome. Moreover, restoration of the NBD1 cotranslational folding defect by second site suppressor mutations also partially restores folding of full-length CFTR. These findings demonstrate that nascent folding intermediates can play an important role in disease pathogenesis and thus provide potential targets for pharmacological correction.
Conflict of interest statement
H.S., J.Y.S., and W.R.S. are employees of the Cystic Fibrosis Foundation, which is involved in and funding independent programs for CF drug development including CFTR modulators that improve CFTR folding. The authors declare no personal competing financial and non-financial interests with Vertex Pharmaceuticals, but acknowledge that as a result of financial support, CFF may be entitled to receive royalty payments from Vertex for marketed products.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
