

ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation
- PMID: 32848148
- PMCID: PMC7449963
- DOI: 10.1038/s41467-020-18035-1
ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation
Abstract
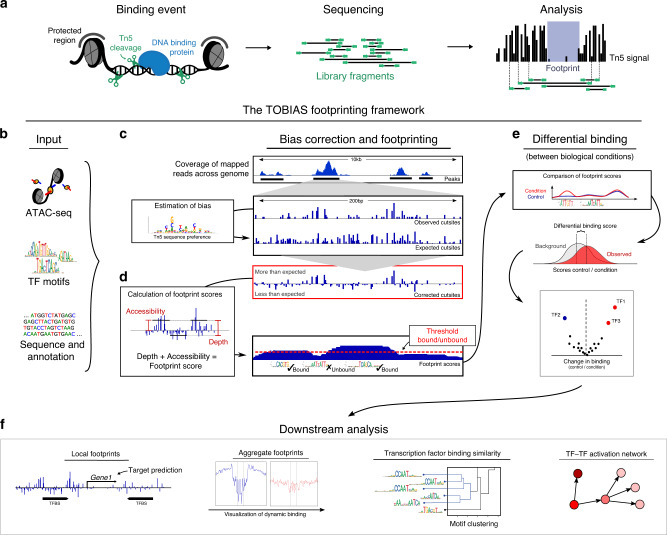
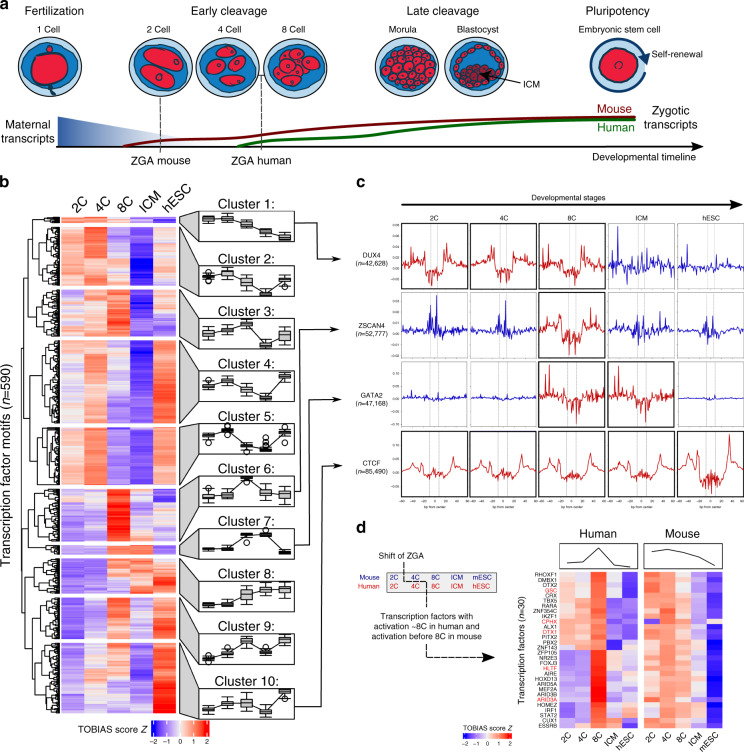
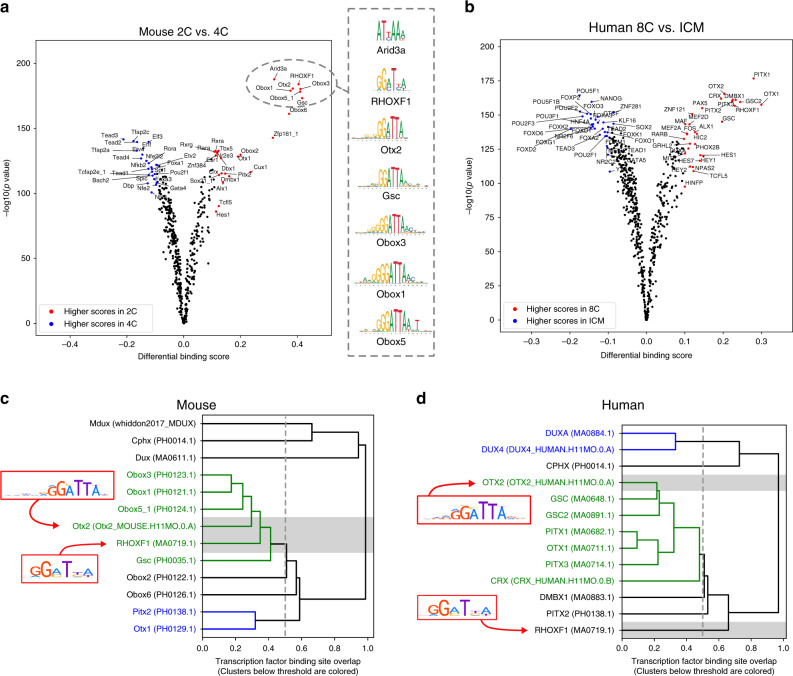
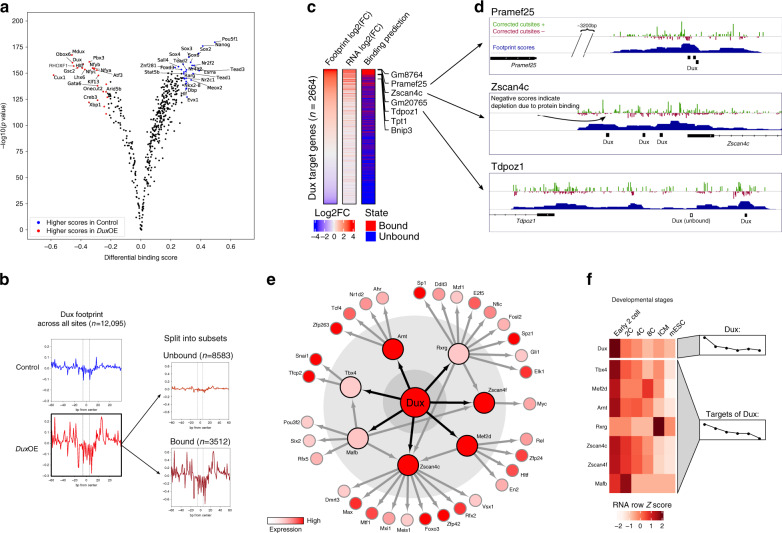
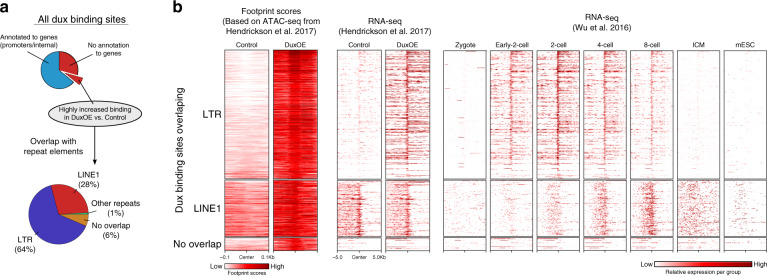
While footprinting analysis of ATAC-seq data can theoretically enable investigation of transcription factor (TF) binding, the lack of a computational tool able to conduct different levels of footprinting analysis has so-far hindered the widespread application of this method. Here we present TOBIAS, a comprehensive, accurate, and fast footprinting framework enabling genome-wide investigation of TF binding dynamics for hundreds of TFs simultaneously. We validate TOBIAS using paired ATAC-seq and ChIP-seq data, and find that TOBIAS outperforms existing methods for bias correction and footprinting. As a proof-of-concept, we illustrate how TOBIAS can unveil complex TF dynamics during zygotic genome activation in both humans and mice, and propose how zygotic Dux activates cascades of TFs, binds to repeat elements and induces expression of novel genetic elements.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
