

LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness
- PMID: 32848593
- PMCID: PMC7419697
- DOI: 10.3389/fnmol.2020.00123
LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness
Abstract
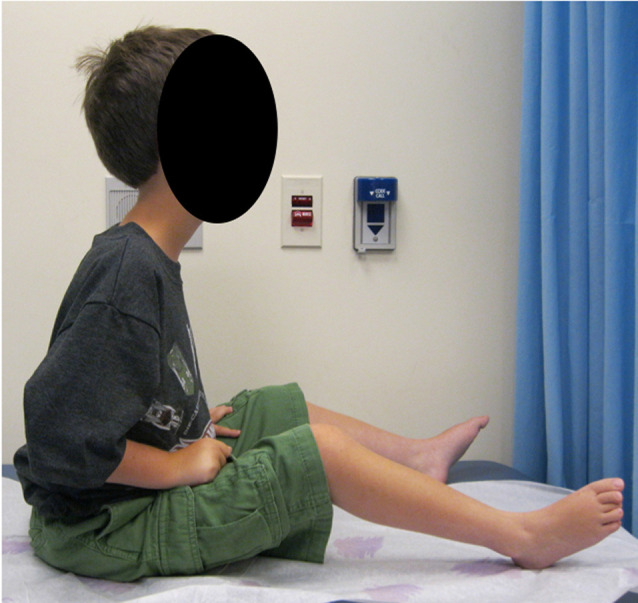
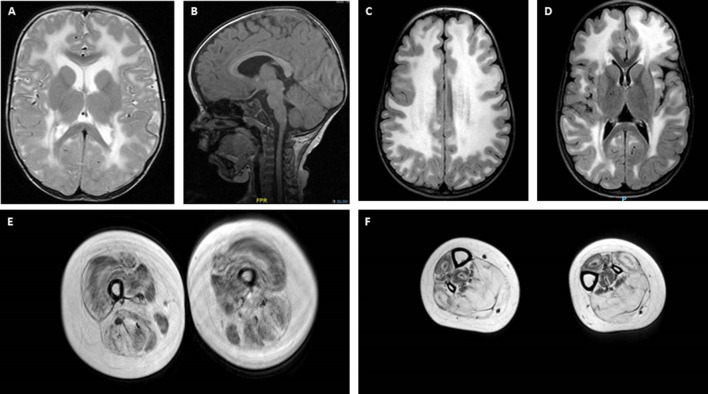
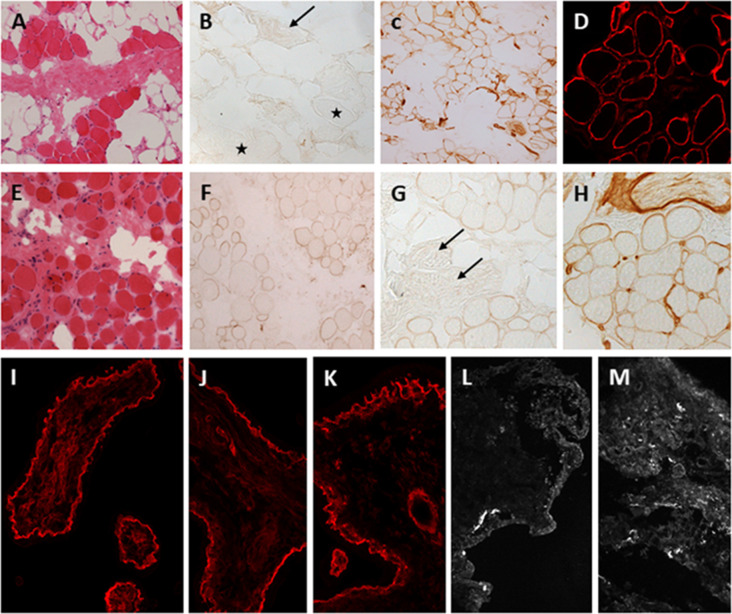
Mutations in the LAMA2 gene affect the production of the α2 subunit of laminin-211 (= merosin) and result in either partial or complete laminin-211 deficiency. Complete merosin deficiency is typically associated with a more severe congenital muscular dystrophy (CMD), clinically manifested by hypotonia and weakness at birth, the development of contractures of large joints, and progressive respiratory involvement. Muscle atrophy and severe weakness typically prevent independent ambulation. Partial merosin deficiency is mostly manifested by later onset limb-girdle weakness and joint contractures so that independent ambulation is typically achieved. Collectively, complete and partial merosin deficiency is referred to as LAMA2-related dystrophies (LAMA2-RDs) and represents one of the most common forms of congenital muscular dystrophies worldwide. LAMA2-RDs are classically characterized by both central and peripheral nervous system involvement with abnormal appearing white matter (WM) on brain MRI and dystrophic appearing muscle on muscle biopsy as well as creatine kinase (CK) levels commonly elevated to >1,000 IU/L. Next-generation sequencing (NGS) has greatly improved diagnostic abilities for LAMA2-RD, and the majority of patients with merosin deficiency carry recessive pathogenic variants in the LAMA2 gene. The existence of multiple animal models for LAMA2-RDs has helped to advance our understanding of laminin-211 and has been instrumental in preclinical research progress and translation to clinical trials. The first clinical trial for the LAMA2-RDs was a phase 1 pharmacokinetic and safety study of the anti-apoptotic compound omigapil, based on preclinical studies performed in the dy W/dy W and dy 2J/dy 2J mouse models. This phase 1 study enabled the collection of pulmonary and motor outcome measures and also provided the opportunity for investigating exploratory outcome measures including muscle ultrasound, muscle MRI and serum, and urine biomarker collection. Natural history studies, including a five-year prospective natural history and comparative outcome measures study in patients with LAMA2-RD, have helped to better delineate the natural history and identify viable outcome measures. Plans for further clinical trials for LAMA2-RDs are presently in progress, highlighting the necessity of identifying adequate, disease-relevant biomarkers, capable of reflecting potential therapeutic changes, in addition to refining the clinical outcome measures and time-to-event trajectory analysis of affected patients.
Keywords: LAMA2; biomarkers; clinical trial; natural history; phenotype.
Copyright © 2020 Sarkozy, Foley, Zambon, Bönnemann and Muntoni.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous