

Targeting Protein Kinase G to Treat Cardiac Proteotoxicity
- PMID: 32848832
- PMCID: PMC7399205
- DOI: 10.3389/fphys.2020.00858
Targeting Protein Kinase G to Treat Cardiac Proteotoxicity
Abstract
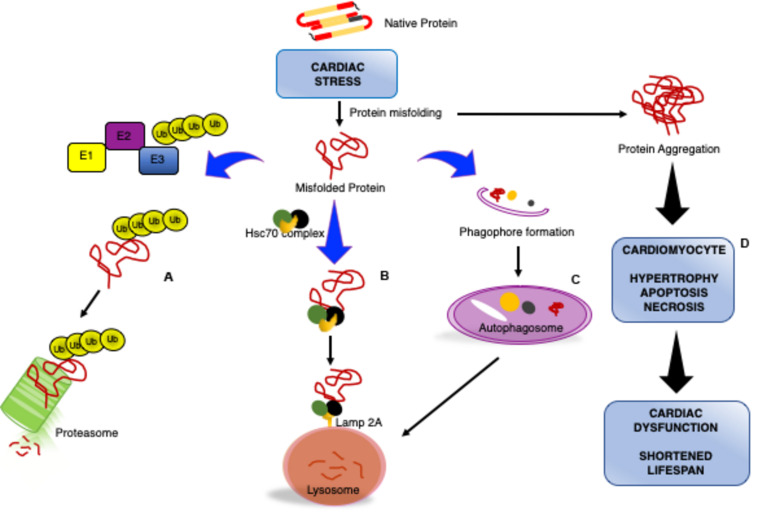
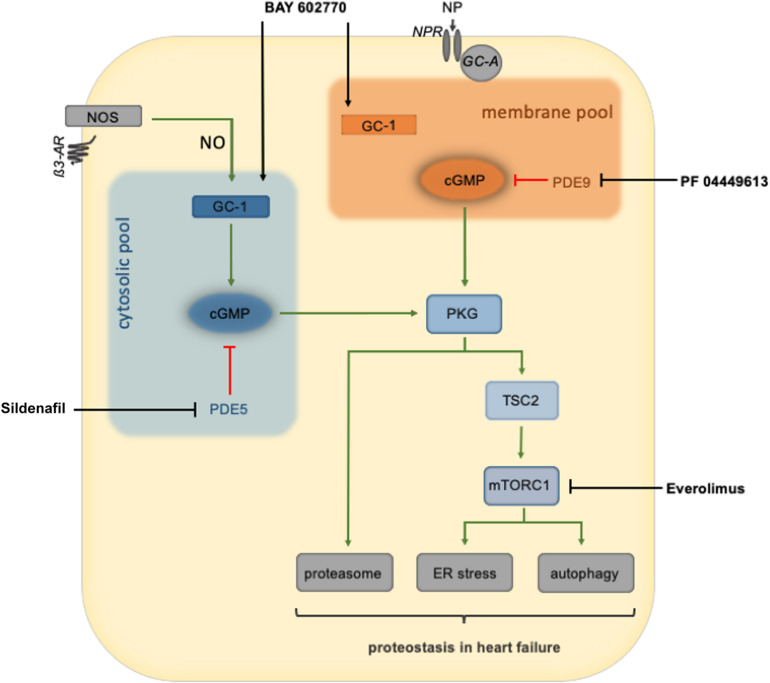
Impaired or insufficient protein kinase G (PKG) signaling and protein quality control (PQC) are hallmarks of most forms of cardiac disease, including heart failure. Their dysregulation has been shown to contribute to and exacerbate cardiac hypertrophy and remodeling, reduced cell survival and disease pathogenesis. Enhancement of PKG signaling and PQC are associated with improved cardiac function and survival in many pre-clinical models of heart disease. While many clinically used pharmacological approaches exist to stimulate PKG, there are no FDA-approved therapies to safely enhance cardiomyocyte PQC. The latter is predominantly due to our lack of knowledge and identification of proteins regulating cardiomyocyte PQC. Recently, multiple studies have demonstrated that PKG regulates PQC in the heart, both during physiological and pathological states. These studies tested already FDA-approved pharmacological therapies to activate PKG, which enhanced cardiomyocyte PQC and alleviated cardiac disease. This review examines the roles of PKG and PQC during disease pathogenesis and summarizes the experimental and clinical data supporting the utility of stimulating PKG to target cardiac proteotoxicity.
Keywords: PKG; autophagy; heart failure; proteasome; proteostasis; proteotoxicity.
Copyright © 2020 Oeing, Mishra, Dunkerly-Eyring and Ranek.
Figures
Similar articles
-
Phosphorylation Modifications Regulating Cardiac Protein Quality Control Mechanisms.Front Physiol. 2020 Nov 12;11:593585. doi: 10.3389/fphys.2020.593585. eCollection 2020. Front Physiol. 2020. PMID: 33281625 Free PMC article. Review.
-
Cellular Protein Quality Control in Diabetic Cardiomyopathy: From Bench to Bedside.Front Cardiovasc Med. 2020 Oct 15;7:585309. doi: 10.3389/fcvm.2020.585309. eCollection 2020. Front Cardiovasc Med. 2020. PMID: 33195472 Free PMC article. Review.
-
Low myocardial protein kinase G activity in heart failure with preserved ejection fraction.Circulation. 2012 Aug 14;126(7):830-9. doi: 10.1161/CIRCULATIONAHA.111.076075. Epub 2012 Jul 17. Circulation. 2012. PMID: 22806632
-
H- ras deletion protects against angiotensin II-induced arterial hypertension and cardiac remodeling through protein kinase G-Iβ pathway activation.FASEB J. 2018 Feb;32(2):920-934. doi: 10.1096/fj.201700134RRRR. Epub 2018 Jan 4. FASEB J. 2018. PMID: 29054855
-
Reviving the protein quality control system: therapeutic target for cardiac disease in the elderly.Trends Cardiovasc Med. 2015 Apr;25(3):243-7. doi: 10.1016/j.tcm.2014.10.013. Epub 2014 Oct 28. Trends Cardiovasc Med. 2015. PMID: 25528995 Review.
Cited by
-
Phosphorylation Modifications Regulating Cardiac Protein Quality Control Mechanisms.Front Physiol. 2020 Nov 12;11:593585. doi: 10.3389/fphys.2020.593585. eCollection 2020. Front Physiol. 2020. PMID: 33281625 Free PMC article. Review.
-
Sacubitril/valsartan reduces proteasome activation and cardiomyocyte area in an experimental mouse model of hypertrophy.J Mol Cell Cardiol Plus. 2024 Jan 7;7:100059. doi: 10.1016/j.jmccpl.2023.100059. eCollection 2024 Mar. J Mol Cell Cardiol Plus. 2024. PMID: 39802437 Free PMC article.
-
Advances, Perspectives and Potential Engineering Strategies of Light-Gated Phosphodiesterases for Optogenetic Applications.Int J Mol Sci. 2020 Oct 13;21(20):7544. doi: 10.3390/ijms21207544. Int J Mol Sci. 2020. PMID: 33066112 Free PMC article. Review.
-
Vps4a Regulates Autophagic Flux to Prevent Hypertrophic Cardiomyopathy.Int J Mol Sci. 2023 Jun 28;24(13):10800. doi: 10.3390/ijms241310800. Int J Mol Sci. 2023. PMID: 37445978 Free PMC article.
-
Cullin Deneddylation Suppresses the Necroptotic Pathway in Cardiomyocytes.Front Physiol. 2021 Jun 28;12:690423. doi: 10.3389/fphys.2021.690423. eCollection 2021. Front Physiol. 2021. PMID: 34262479 Free PMC article.
References
-
- Armstrong P. W., Roessig L., Patel M. J., Anstrom K. J., Butler J., Voors A. A., et al. (2018). A multicenter, randomized, double-blind, placebo-controlled trial of the efficacy and safety of the oral soluble guanylate cyclase stimulator: the VICTORIA trial. JACC Heart Fail. 6 96–104. 10.1016/j.jchf.2017.08.013 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources