

Analytical Performance Validation of Next-Generation Sequencing Based Clinical Microbiology Assays Using a K-mer Analysis Workflow
- PMID: 32849463
- PMCID: PMC7422695
- DOI: 10.3389/fmicb.2020.01883
Analytical Performance Validation of Next-Generation Sequencing Based Clinical Microbiology Assays Using a K-mer Analysis Workflow
Abstract
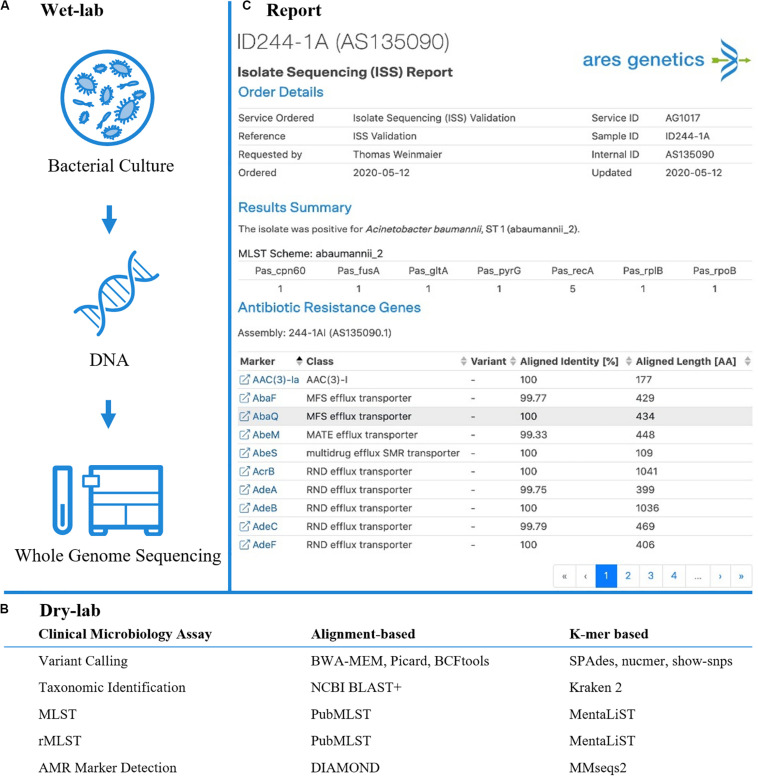
Next-generation sequencing (NGS) enables clinical microbiology assays such as molecular typing of bacterial isolates which is now routinely applied for infection control and epidemiology. Additionally, feasibility for NGS-based identification of antimicrobial resistance (AMR) markers as well as genetic prediction of antibiotic susceptibility testing results has been demonstrated. Various bioinformatics approaches enabling NGS-based clinical microbiology assays exist, but standardized, computationally efficient and scalable sample-to-results workflows including validated quality control parameters are still lacking. Bioinformatics analysis workflows based on k-mers have been shown to allow for fast and efficient analysis of large genomics data sets as obtained from microbial sequencing applications. We here demonstrate applicability of k-mer based clinical microbiology assays for whole-genome sequencing (WGS) including variant calling, taxonomic identification, bacterial typing as well as AMR marker detection. The wet-lab and dry-lab workflows were developed and validated in line with Clinical Laboratory Improvement Act (CLIA) guidelines for laboratory-developed tests (LDTs) on multi-drug resistant ESKAPE pathogens. The developed k-mer based workflow demonstrated ≥99.39% repeatability, ≥99.09% reproducibility and ≥99.76% accuracy for variant calling and applied assays as determined by intra-day and inter-day triplicate measurements. The limit of detection (LOD) across assays was found to be at 20× sequencing depth and 15× for AMR marker detection. Thorough benchmarking of the k-mer based workflow revealed analytical performance criteria are comparable to state-of-the-art alignment based workflows across clinical microbiology assays. Diagnostic sensitivity and specificity for multilocus sequence typing (MLST) and phylogenetic analysis were 100% for both approaches. For AMR marker detection, sensitivity and specificity were 95.29 and 99.78% for the k-mer based workflow as compared to 95.17 and 99.77% for the alignment-based approach. Summarizing, results illustrate that k-mer based analysis workflows enable a broad range of clinical microbiology assays, potentially not only for WGS-based typing and AMR gene detection but also genetic prediction of antibiotic susceptibility testing results.
Keywords: antimicrobial resistance; human pathogens; k-mer analysis; whole genome sequencing; workflow validation.
Copyright © 2020 Lepuschitz, Weinmaier, Mrazek, Beisken, Weinberger and Posch.
Figures
References
-
- Bogaerts B., Winand R., Fu Q., Van Braekel J., Ceyssens P.-J., Mattheus W., et al. (2019). Validation of a Bioinformatics Workflow for Routine Analysis of Whole-Genome Sequencing Data and Related Challenges for Pathogen Typing in a European National Reference Center: Neisseria meningitidis as a Proof-of-Concept. Front. Microbiol. 10:362. 10.3389/fmicb.2019.00362 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous