

Contribution of mRNA Splicing to Mismatch Repair Gene Sequence Variant Interpretation
- PMID: 32849802
- PMCID: PMC7398121
- DOI: 10.3389/fgene.2020.00798
Contribution of mRNA Splicing to Mismatch Repair Gene Sequence Variant Interpretation
Abstract
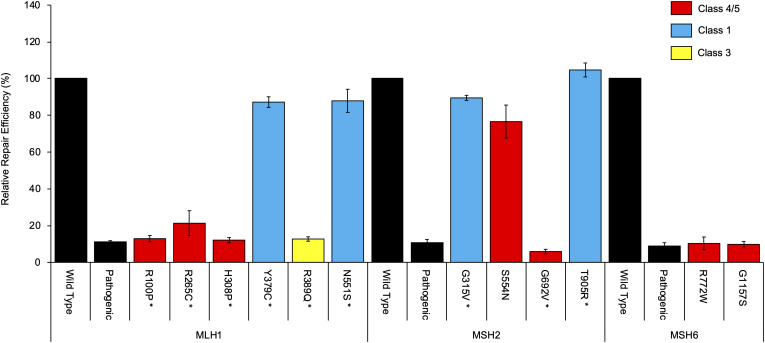
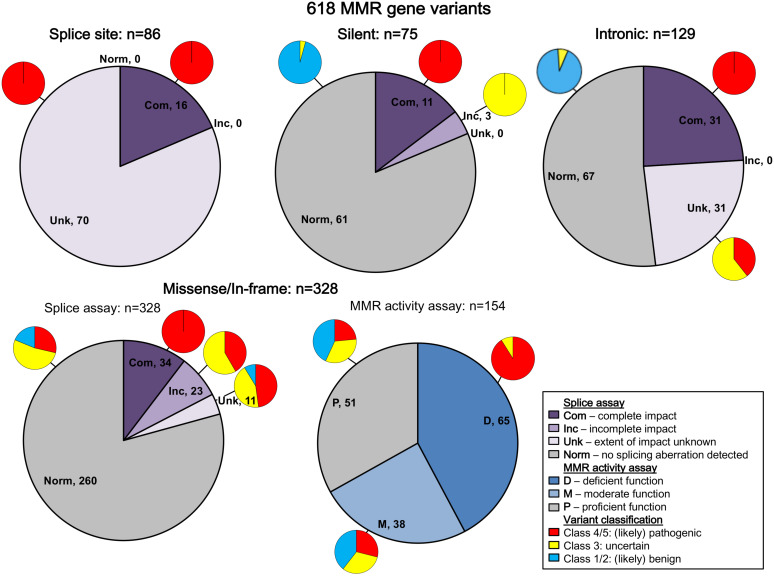
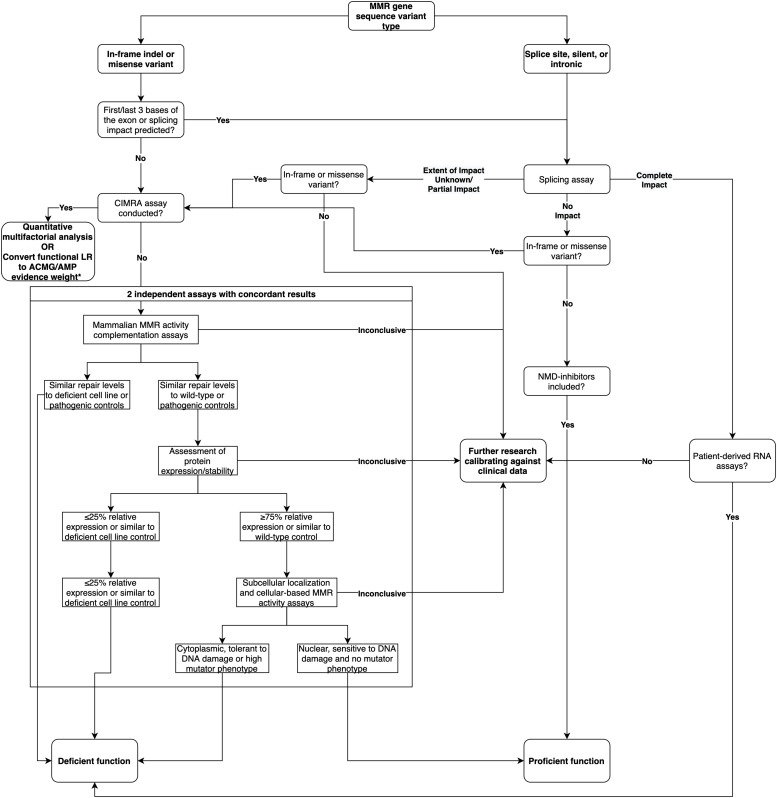
Functional assays that assess mRNA splicing can be used in interpretation of the clinical significance of sequence variants, including the Lynch syndrome-associated mismatch repair (MMR) genes. The purpose of this study was to investigate the contribution of splicing assay data to the classification of MMR gene sequence variants. We assayed mRNA splicing for 24 sequence variants in MLH1, MSH2, and MSH6, including 12 missense variants that were also assessed using a cell-free in vitro MMR activity (CIMRA) assay. Multifactorial likelihood analysis was conducted for each variant, combining CIMRA outputs and clinical data where available. We collated these results with existing public data to provide a dataset of splicing assay results for a total of 671 MMR gene sequence variants (328 missense/in-frame indel), and published and unpublished repair activity measurements for 154 of these variants. There were 241 variants for which a splicing aberration was detected: 92 complete impact, 33 incomplete impact, and 116 where it was not possible to determine complete versus incomplete splicing impact. Splicing results mostly aided in the interpretation of intronic (72%) and silent (92%) variants and were the least useful for missense substitutions/in-frame indels (10%). MMR protein functional activity assays were more useful in the analysis of these exonic variants but by design they were not able to detect clinically important splicing aberrations identified by parallel mRNA assays. The development of high throughput assays that can quantitatively assess impact on mRNA transcript expression and protein function in parallel will streamline classification of MMR gene sequence variants.
Keywords: Lynch syndrome; mRNA splicing; mismatch repair genes; splicing aberrations; variant interpretation and classification; variant type.
Copyright © 2020 Thompson, Walters, Parsons, Dumenil, Drost, Tiersma, Lindor, Tavtigian, de Wind, Spurdle and the InSiGHT Variant Interpretation Committee.
Figures
References
-
- Buchanan D. D., Tan Y. Y., Walsh M. D., Clendenning M., Metcalf A. M., Ferguson K., et al. (2014). Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J. Clin. Oncol. 32 90–100. 10.1200/JCO.2013.51.2129 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous