

Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination
- PMID: 32853178
- PMCID: PMC7526550
- DOI: 10.1172/jci.insight.139983
Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination
Abstract
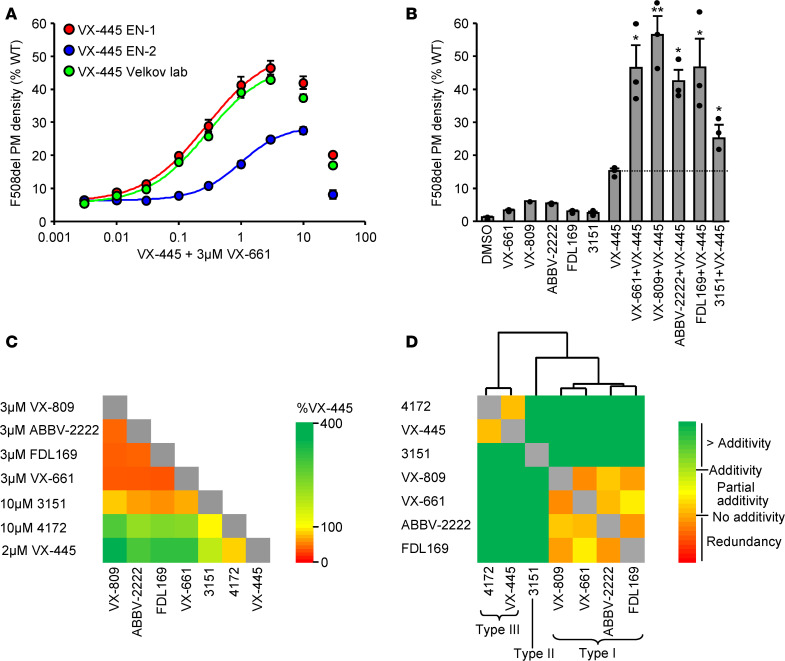
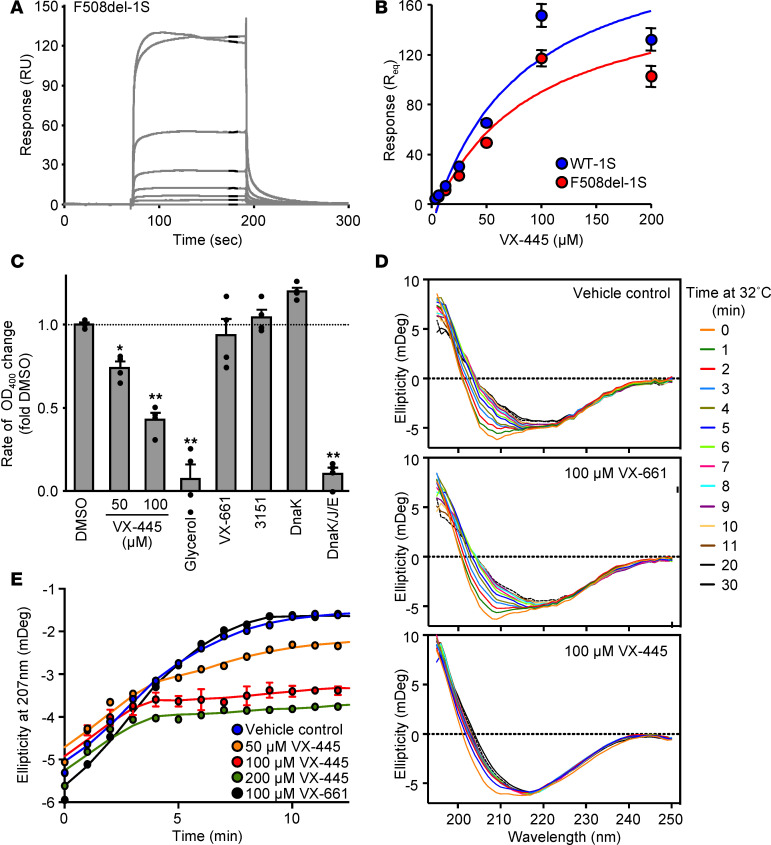
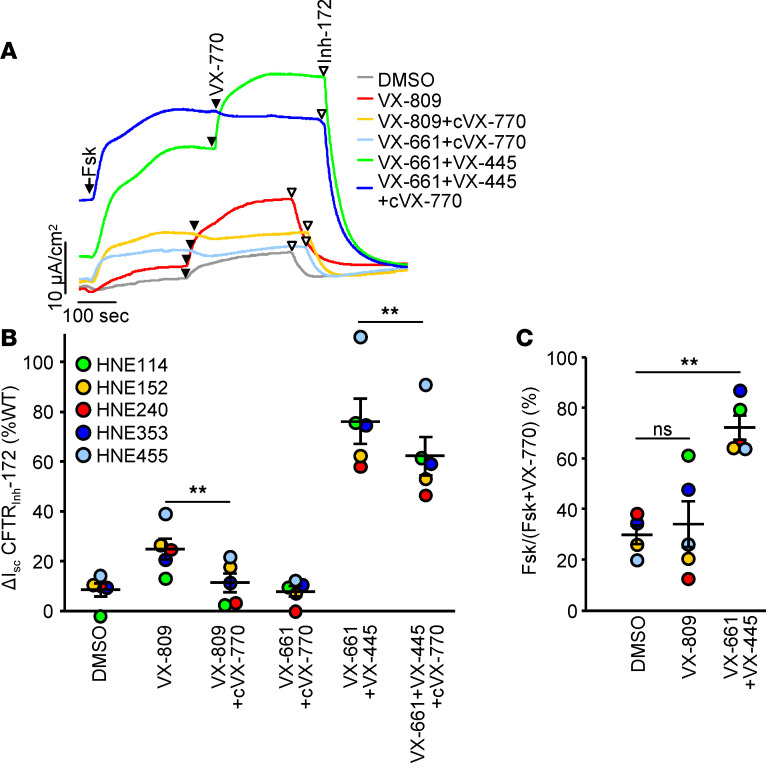
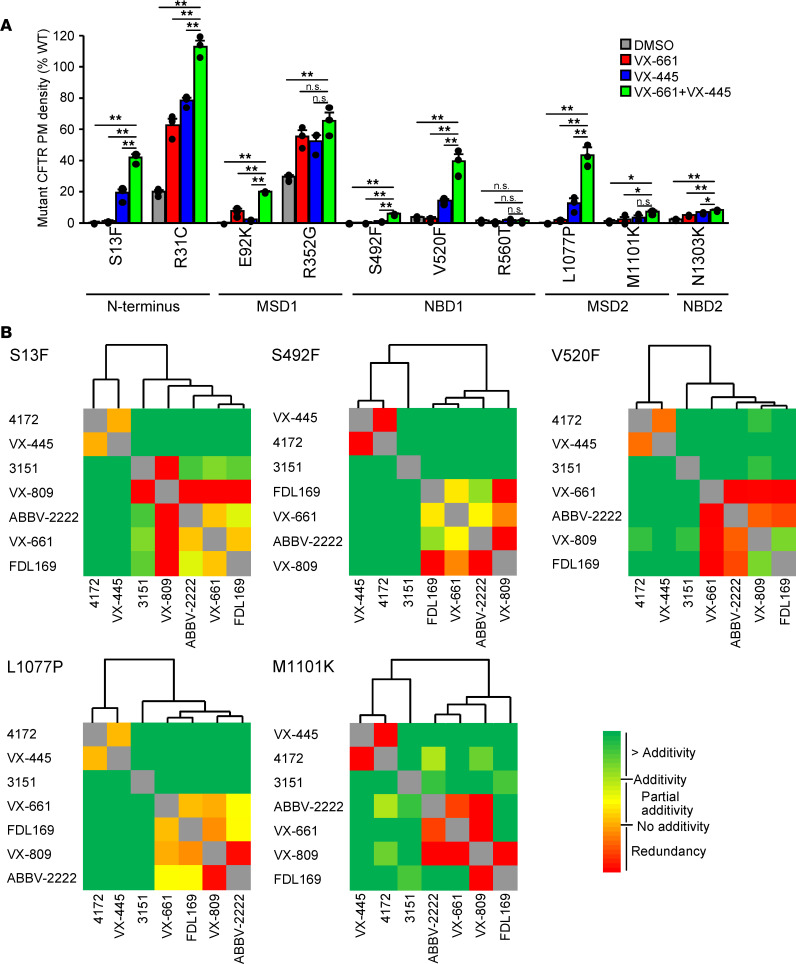
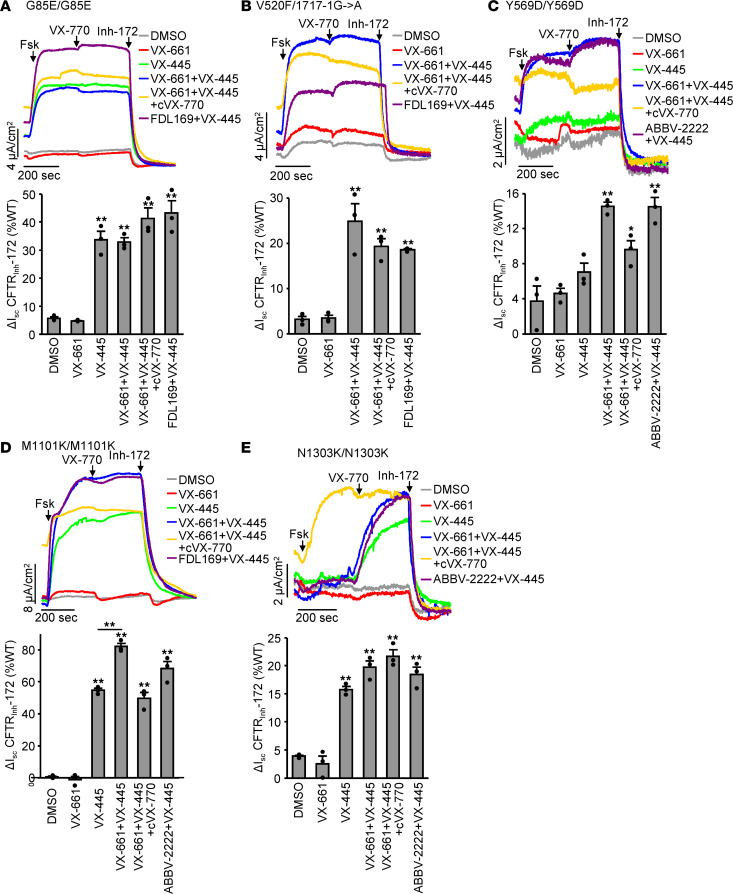
Based on its clinical benefits, Trikafta - the combination of folding correctors VX-661 (tezacaftor), VX-445 (elexacaftor), and the gating potentiator VX-770 (ivacaftor) - was FDA approved for treatment of patients with cystic fibrosis (CF) carrying deletion of phenylalanine at position 508 (F508del) of the CF transmembrane conductance regulator (CFTR) on at least 1 allele. Neither the mechanism of action of VX-445 nor the susceptibility of rare CF folding mutants to Trikafta are known. Here, we show that, in human bronchial epithelial cells, VX-445 synergistically restores F508del-CFTR processing in combination with type I or II correctors that target the nucleotide binding domain 1 (NBD1) membrane spanning domains (MSDs) interface and NBD2, respectively, consistent with a type III corrector mechanism. This inference was supported by the VX-445 binding to and unfolding suppression of the isolated F508del-NBD1 of CFTR. The VX-661 plus VX-445 treatment restored F508del-CFTR chloride channel function in the presence of VX-770 to approximately 62% of WT CFTR in homozygous nasal epithelia. Substantial rescue of rare misprocessing mutations (S13F, R31C, G85E, E92K, V520F, M1101K, and N1303K), confined to MSD1, MSD2, NBD1, and NBD2 of CFTR, was also observed in airway epithelia, suggesting an allosteric correction mechanism and the possible application of Trikafta for patients with rare misfolding mutants of CFTR.
Keywords: Cell Biology; Chloride channels; Drug therapy; Epithelial transport of ions and water; Pulmonology.
Conflict of interest statement
Figures
Similar articles
-
Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment.Int J Mol Sci. 2021 May 17;22(10):5262. doi: 10.3390/ijms22105262. Int J Mol Sci. 2021. PMID: 34067708 Free PMC article.
-
Targeting the E1 ubiquitin-activating enzyme (UBA1) improves elexacaftor/tezacaftor/ivacaftor efficacy towards F508del and rare misfolded CFTR mutants.Cell Mol Life Sci. 2022 Mar 16;79(4):192. doi: 10.1007/s00018-022-04215-3. Cell Mol Life Sci. 2022. PMID: 35292885 Free PMC article.
-
The rescue of F508del-CFTR by elexacaftor/tezacaftor/ivacaftor (Trikafta) in human airway epithelial cells is underestimated due to the presence of ivacaftor.Eur Respir J. 2022 Feb 24;59(2):2100671. doi: 10.1183/13993003.00671-2021. Print 2022 Feb. Eur Respir J. 2022. PMID: 34266939
-
Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del).Cochrane Database Syst Rev. 2020 Dec 17;12(12):CD010966. doi: 10.1002/14651858.CD010966.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2023 Nov 20;11:CD010966. doi: 10.1002/14651858.CD010966.pub4. PMID: 33331662 Free PMC article. Updated.
-
The preclinical discovery and development of the combination of ivacaftor + tezacaftor used to treat cystic fibrosis.Expert Opin Drug Discov. 2020 Aug;15(8):873-891. doi: 10.1080/17460441.2020.1750592. Epub 2020 Apr 15. Expert Opin Drug Discov. 2020. PMID: 32290721 Review.
Cited by
-
Allosteric inhibition of CFTR gating by CFTRinh-172 binding in the pore.Nat Commun. 2024 Aug 6;15(1):6668. doi: 10.1038/s41467-024-50641-1. Nat Commun. 2024. PMID: 39107303 Free PMC article.
-
Measurements of spontaneous CFTR-mediated ion transport without acute channel activation in airway epithelial cultures after modulator exposure.Sci Rep. 2021 Nov 19;11(1):22616. doi: 10.1038/s41598-021-02044-1. Sci Rep. 2021. PMID: 34799640 Free PMC article.
-
New Therapies to Correct the Cystic Fibrosis Basic Defect.Int J Mol Sci. 2021 Jun 8;22(12):6193. doi: 10.3390/ijms22126193. Int J Mol Sci. 2021. PMID: 34201249 Free PMC article. Review.
-
BOS-318 treatment enhances elexacaftor-tezacaftor-ivacaftor-mediated improvements in airway hydration and mucociliary transport.ERJ Open Res. 2025 Feb 25;11(1):00445-2024. doi: 10.1183/23120541.00445-2024. eCollection 2025 Jan. ERJ Open Res. 2025. PMID: 40013020 Free PMC article.
-
Esc peptides and derivatives potentiate the activity of CFTR with gating defects and display antipseudomonal activity in cystic fibrosis-like lung disease.Cell Mol Life Sci. 2025 Mar 18;82(1):121. doi: 10.1007/s00018-025-05633-9. Cell Mol Life Sci. 2025. PMID: 40100363 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
