

Expanding the space of protein geometries by computational design of de novo fold families
- PMID: 32855341
- PMCID: PMC7787817
- DOI: 10.1126/science.abc0881
Expanding the space of protein geometries by computational design of de novo fold families
Abstract
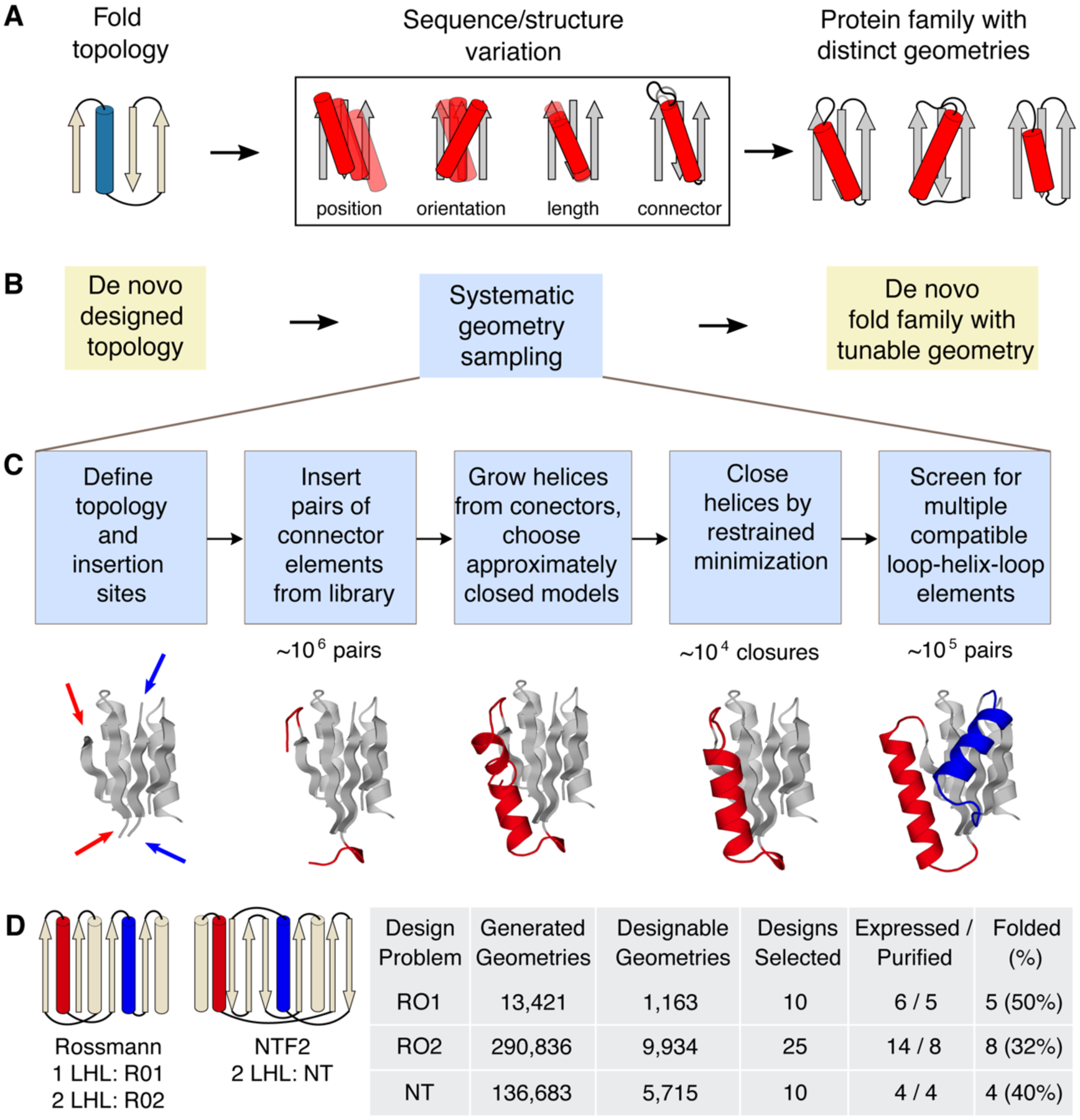
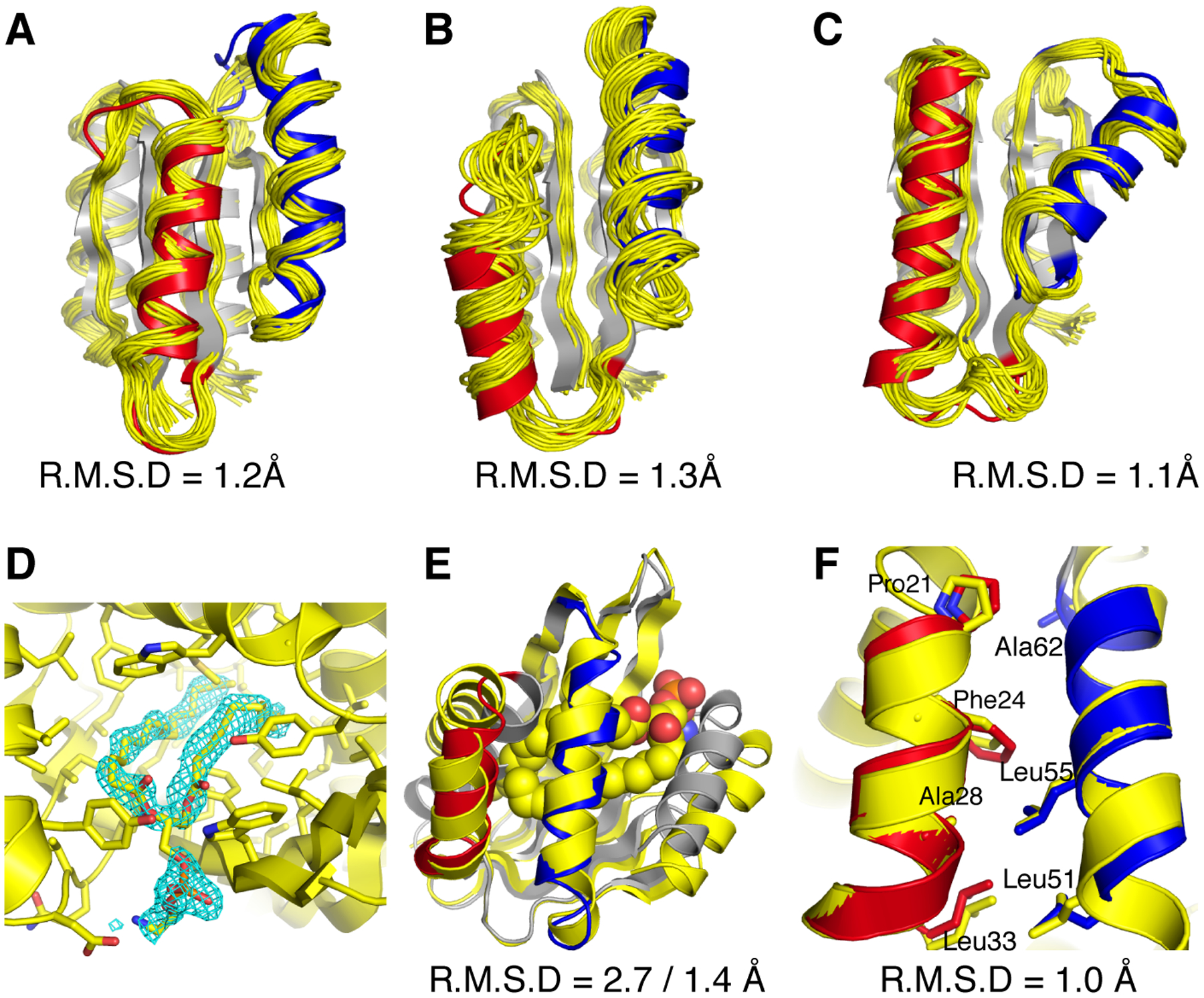
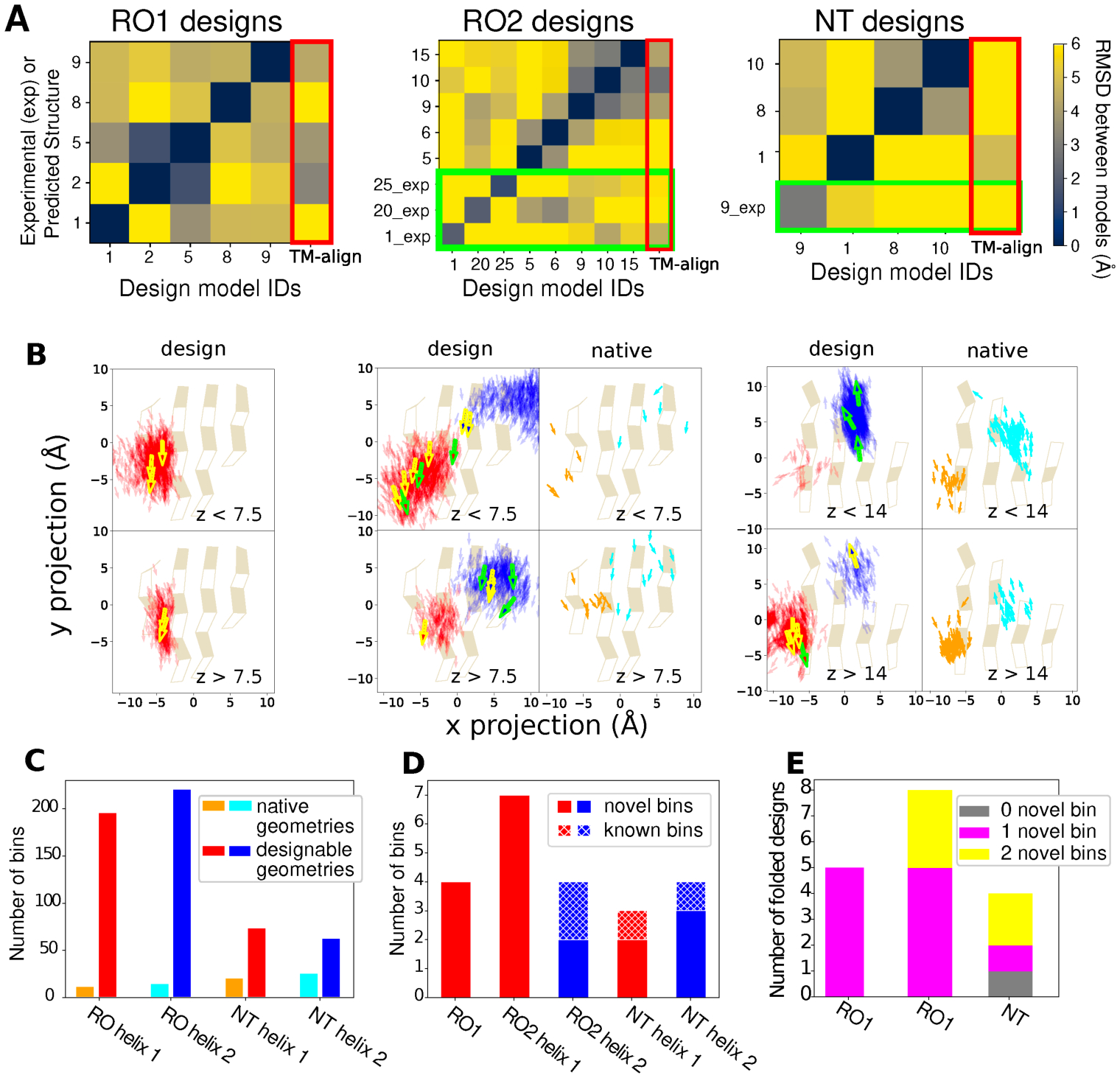
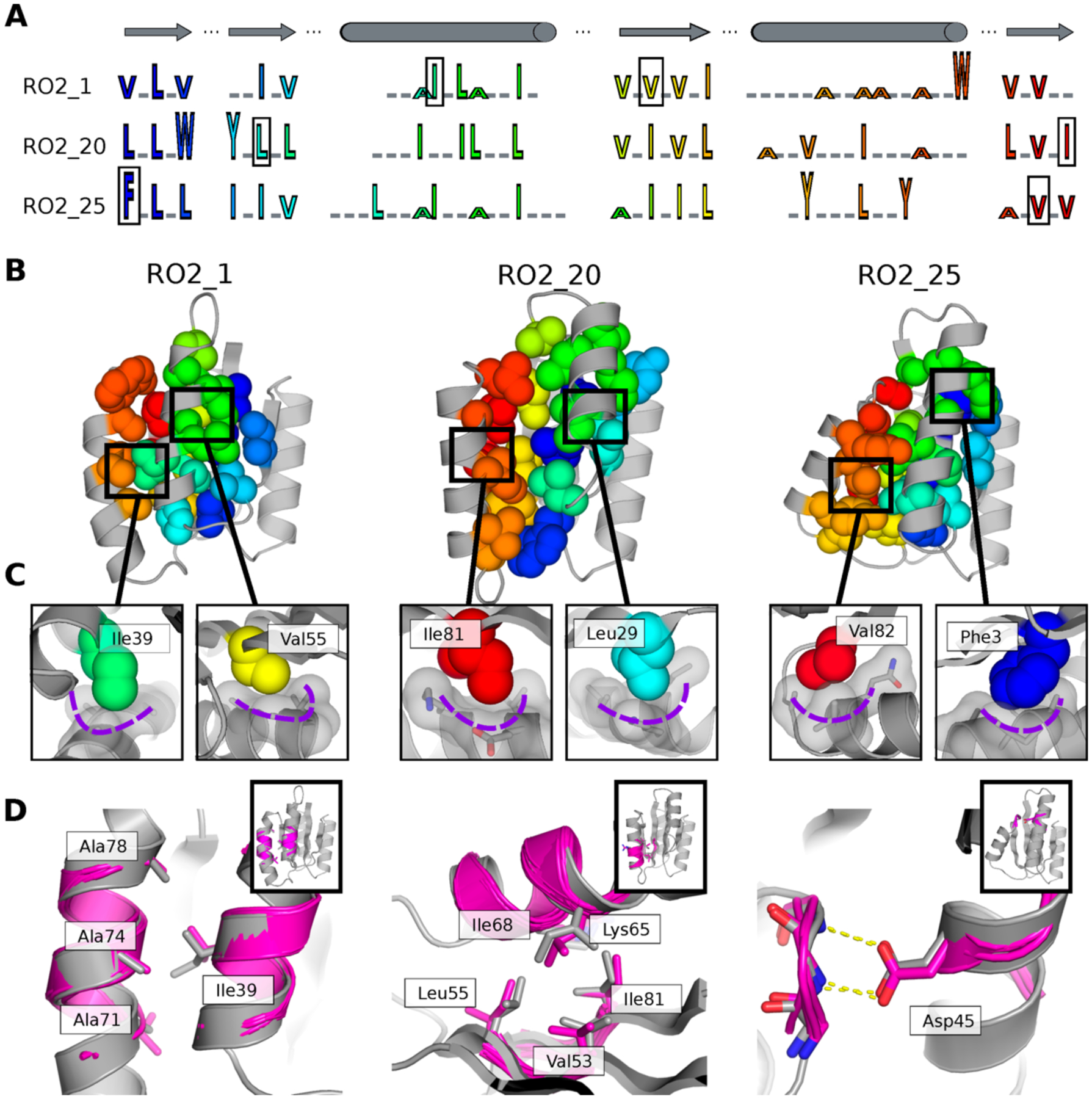
Naturally occurring proteins vary the precise geometries of structural elements to create distinct shapes optimal for function. We present a computational design method, loop-helix-loop unit combinatorial sampling (LUCS), that mimics nature's ability to create families of proteins with the same overall fold but precisely tunable geometries. Through near-exhaustive sampling of loop-helix-loop elements, LUCS generates highly diverse geometries encompassing those found in nature but also surpassing known structure space. Biophysical characterization showed that 17 (38%) of 45 tested LUCS designs encompassing two different structural topologies were well folded, including 16 with designed non-native geometries. Four experimentally solved structures closely matched the designs. LUCS greatly expands the designable structure space and offers a new paradigm for designing proteins with tunable geometries that may be customizable for novel functions.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Competing interests:
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
