

Arylsulfatase K inactivation causes mucopolysaccharidosis due to deficient glucuronate desulfation of heparan and chondroitin sulfate
- PMID: 32856704
- PMCID: PMC8261863
- DOI: 10.1042/BCJ20200546
Arylsulfatase K inactivation causes mucopolysaccharidosis due to deficient glucuronate desulfation of heparan and chondroitin sulfate
Abstract
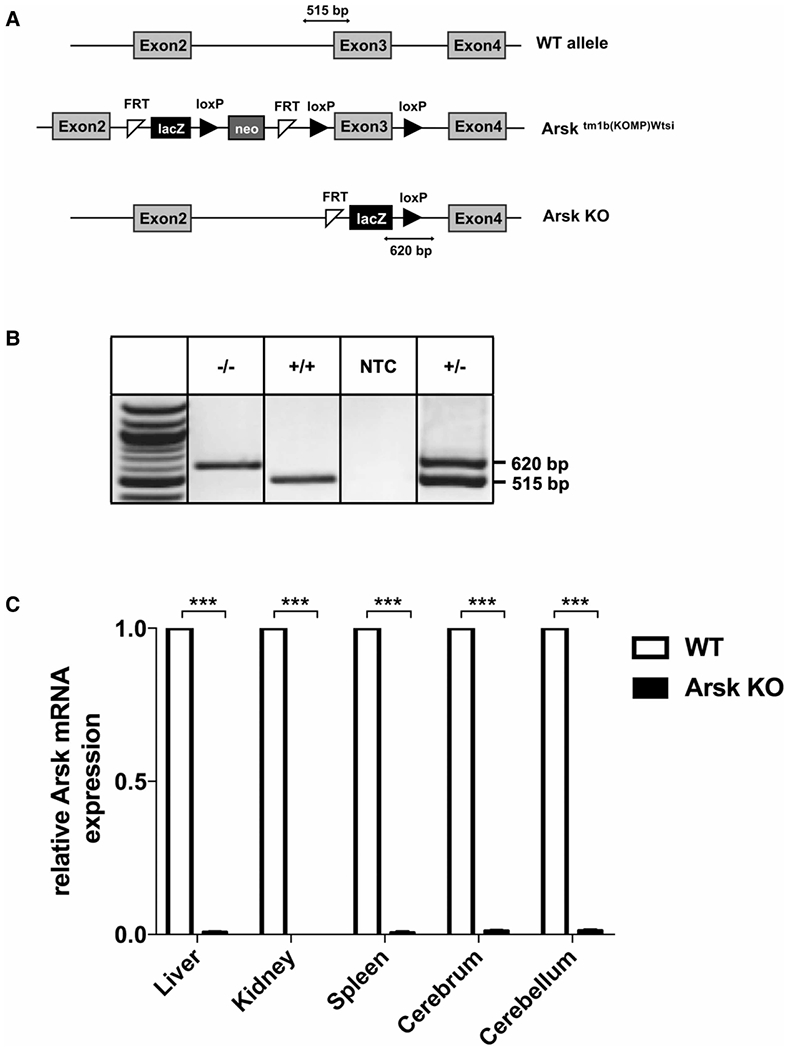
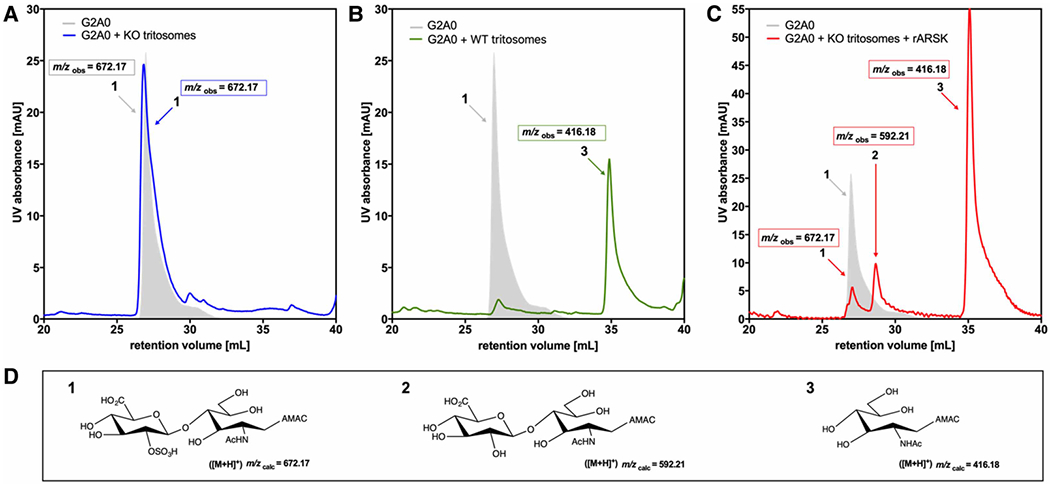
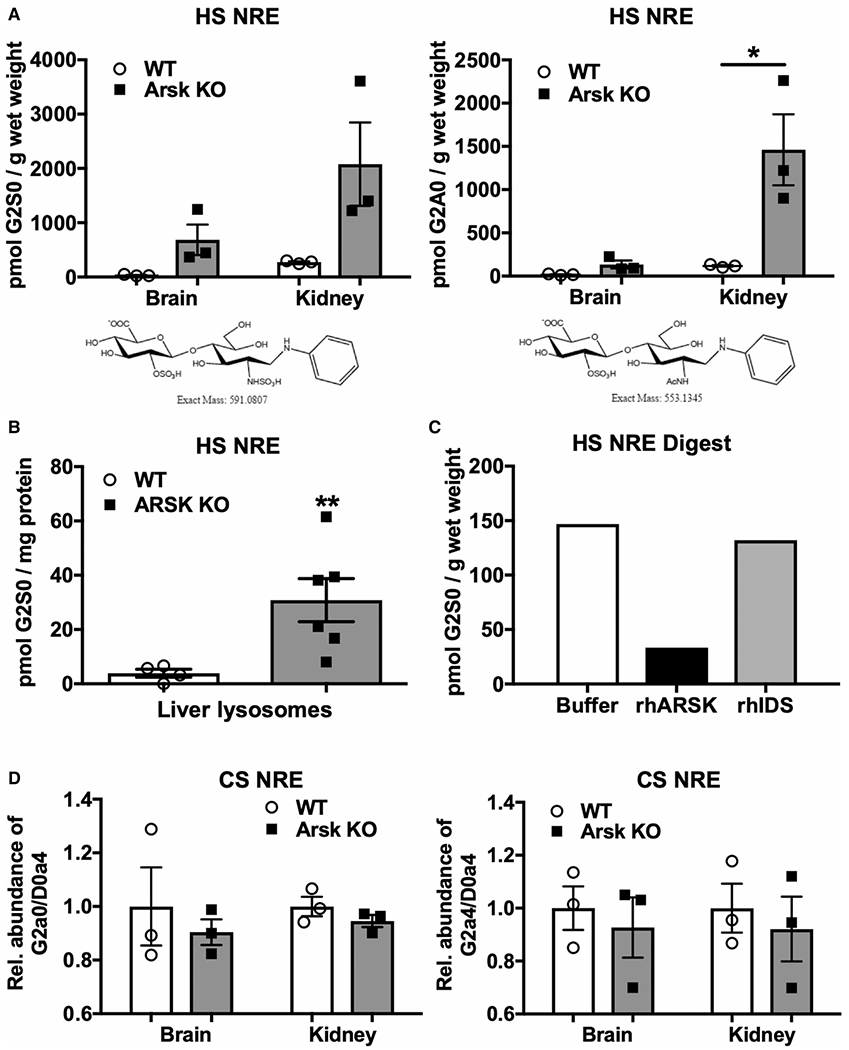
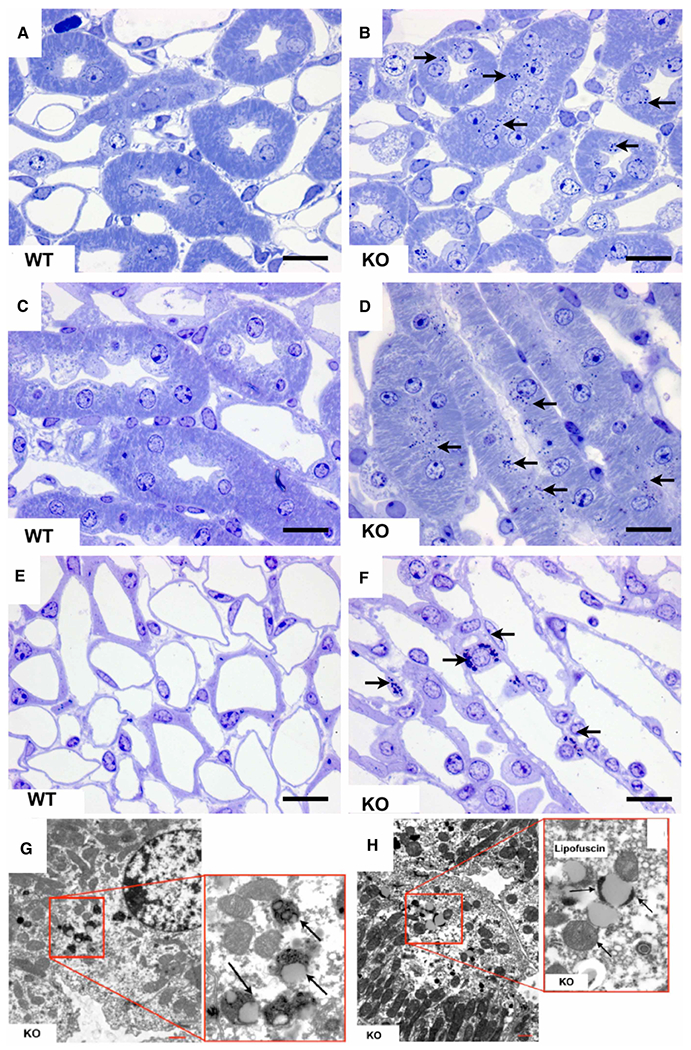
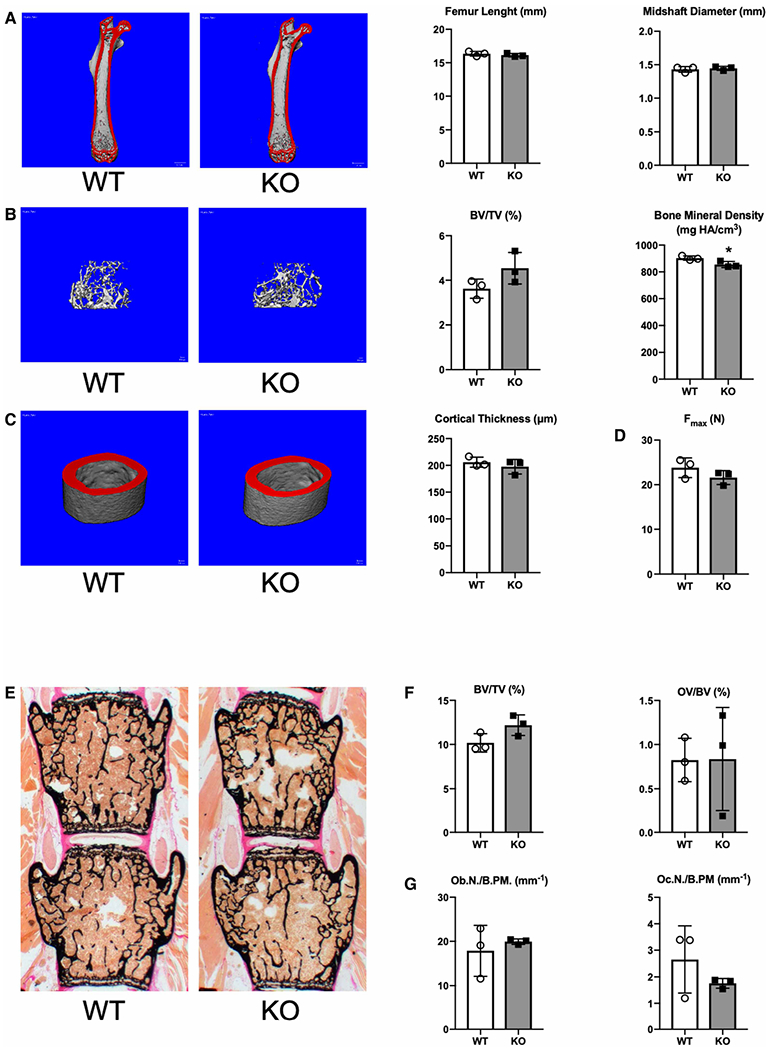
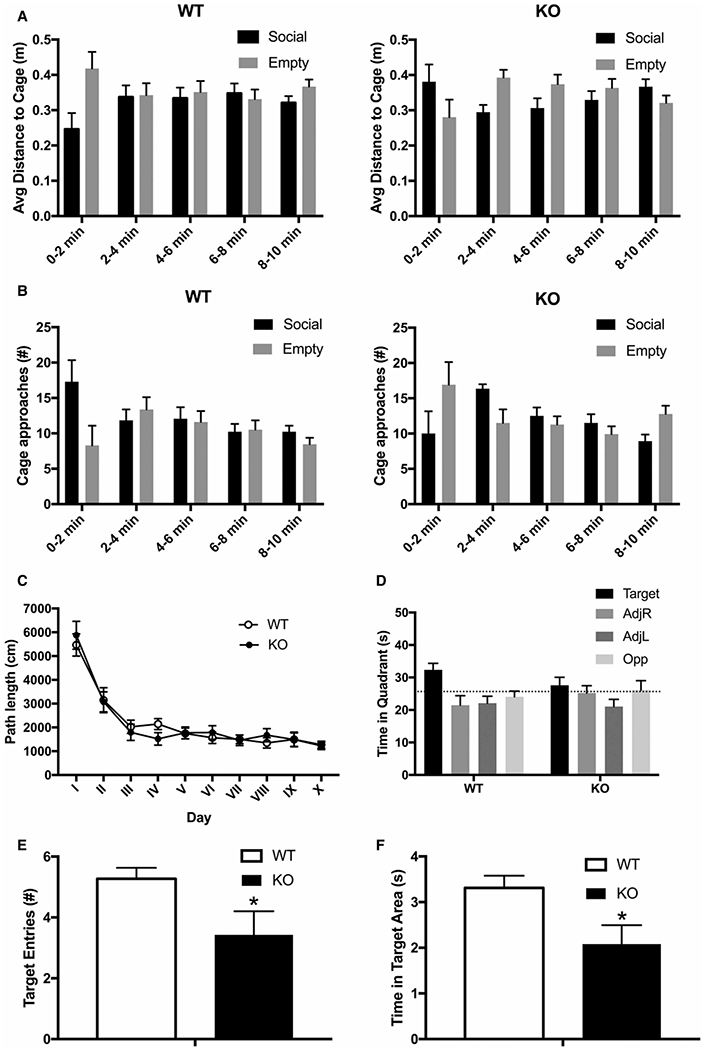
Mucopolysaccharidoses comprise a group of rare metabolic diseases, in which the lysosomal degradation of glycosaminoglycans (GAGs) is impaired due to genetically inherited defects of lysosomal enzymes involved in GAG catabolism. The resulting intralysosomal accumulation of GAG-derived metabolites consequently manifests in neurological symptoms and also peripheral abnormalities in various tissues like liver, kidney, spleen and bone. As each GAG consists of differently sulfated disaccharide units, it needs a specific, but also partly overlapping set of lysosomal enzymes to accomplish their complete degradation. Recently, we identified and characterized the lysosomal enzyme arylsulfatase K (Arsk) exhibiting glucuronate-2-sulfatase activity as needed for the degradation of heparan sulfate (HS), chondroitin sulfate (CS) and dermatan sulfate (DS). In the present study, we investigated the physiological relevance of Arsk by means of a constitutive Arsk knockout mouse model. A complete lack of glucuronate desulfation was demonstrated by a specific enzyme activity assay. Arsk-deficient mice show, in an organ-specific manner, a moderate accumulation of HS and CS metabolites characterized by 2-O-sulfated glucuronate moieties at their non-reducing ends. Pathophysiological studies reflect a rather mild phenotype including behavioral changes. Interestingly, no prominent lysosomal storage pathology like bone abnormalities were detected. Our results from the Arsk mouse model suggest a new although mild form of mucopolysacharidose (MPS), which we designate MPS type IIB.
Keywords: desulfation; glycosaminoglycan degradation; lysosomal storage disorders; lysosomal sulfatases; mucopolysaccharidosis.
© 2020 The Author(s). Published by Portland Press Limited on behalf of the Biochemical Society.
Conflict of interest statement
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Figures

Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Neuraminidase 1 secondary deficiency contributes to CNS pathology in neurological mucopolysaccharidoses via brain protein hypersialylation.J Clin Invest. 2025 Jun 17;135(16):e177430. doi: 10.1172/JCI177430. eCollection 2025 Aug 15. J Clin Invest. 2025. PMID: 40540388 Free PMC article.
-
Novel subtype of mucopolysaccharidosis caused by arylsulfatase K (ARSK) deficiency.J Med Genet. 2022 Oct;59(10):957-964. doi: 10.1136/jmedgenet-2021-108061. Epub 2021 Dec 16. J Med Genet. 2022. PMID: 34916232 Free PMC article.
-
Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome).Cochrane Database Syst Rev. 2016 Feb 5;2(2):CD008185. doi: 10.1002/14651858.CD008185.pub4. Cochrane Database Syst Rev. 2016. PMID: 26845288 Free PMC article.
-
Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome).Cochrane Database Syst Rev. 2014 Jan 8;(1):CD008185. doi: 10.1002/14651858.CD008185.pub3. Cochrane Database Syst Rev. 2014. Update in: Cochrane Database Syst Rev. 2016 Feb 05;2:CD008185. doi: 10.1002/14651858.CD008185.pub4. PMID: 24399699 Updated.
Cited by
-
Endogenous, non-reducing end glycosaminoglycan biomarkers are superior to internal disaccharide glycosaminoglycan biomarkers for newborn screening of mucopolysaccharidoses and GM1 gangliosidosis.Mol Genet Metab. 2023 Sep-Oct;140(1-2):107632. doi: 10.1016/j.ymgme.2023.107632. Epub 2023 Jun 24. Mol Genet Metab. 2023. PMID: 37407323 Free PMC article.
-
A novel homozygous missense variant in ARSK causes MPS X, a new subtype of mucopolysaccharidosis.Genes Dis. 2023 Jul 10;11(3):101025. doi: 10.1016/j.gendis.2023.06.003. eCollection 2024 May. Genes Dis. 2023. PMID: 38292179 Free PMC article. No abstract available.
-
Metabolic Cardiomyopathies and Cardiac Defects in Inherited Disorders of Carbohydrate Metabolism: A Systematic Review.Int J Mol Sci. 2023 May 11;24(10):8632. doi: 10.3390/ijms24108632. Int J Mol Sci. 2023. PMID: 37239976 Free PMC article.
-
Misdiagnosis in mucopolysaccharidoses.J Appl Genet. 2022 Sep;63(3):475-495. doi: 10.1007/s13353-022-00703-1. Epub 2022 May 13. J Appl Genet. 2022. PMID: 35562626 Review.
-
Mucopolysaccharidosis-Plus Syndrome: Is This a Type of Mucopolysaccharidosis or a Separate Kind of Metabolic Disease?Int J Mol Sci. 2024 Sep 4;25(17):9570. doi: 10.3390/ijms25179570. Int J Mol Sci. 2024. PMID: 39273517 Free PMC article. Review.
References
-
- von Figura K, Gieselmann V and Jaeken J (2001) Metachromatic leukodystrophy. In The Metabolic & Molecular Bases of Inherited Diseases (Scriver CR et al. ed), pp. 3694–3724, McGraw-Hill, New York
-
- Neufeld EF and Muenzer J (2001) The mucopolysaccharidoses. In The Metabolic & Molecular Bases of Inherited Diseases (Scriver CR et al. eds), pp. 3421–3452, McGraw-Hill, New York. Chapter 136
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials