

GSK3-ARC/Arg3.1 and GSK3-Wnt signaling axes trigger amyloid-β accumulation and neuroinflammation in middle-aged Shugoshin 1 mice
- PMID: 32857910
- PMCID: PMC7576275
- DOI: 10.1111/acel.13221
GSK3-ARC/Arg3.1 and GSK3-Wnt signaling axes trigger amyloid-β accumulation and neuroinflammation in middle-aged Shugoshin 1 mice
Abstract
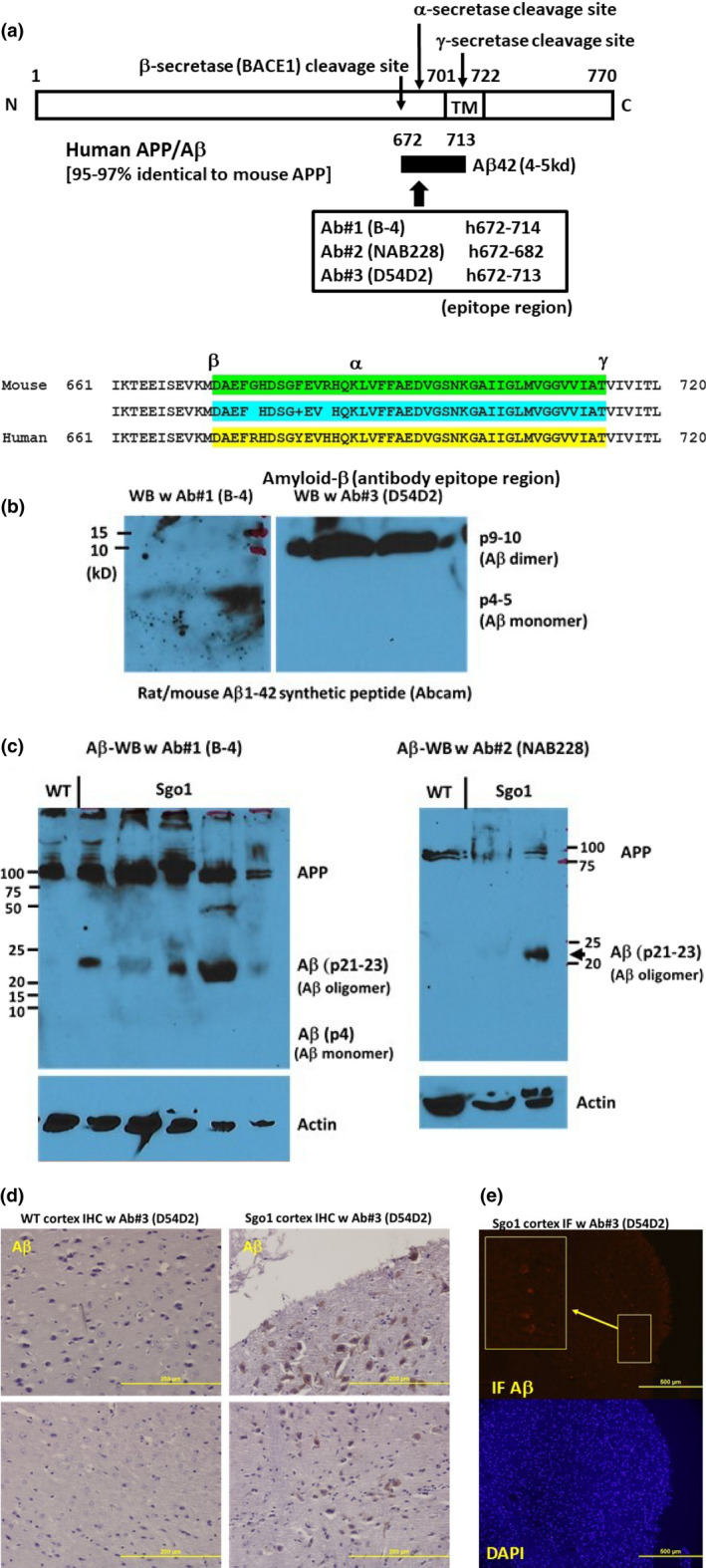
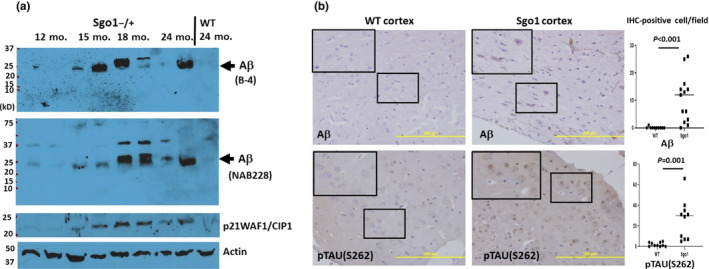
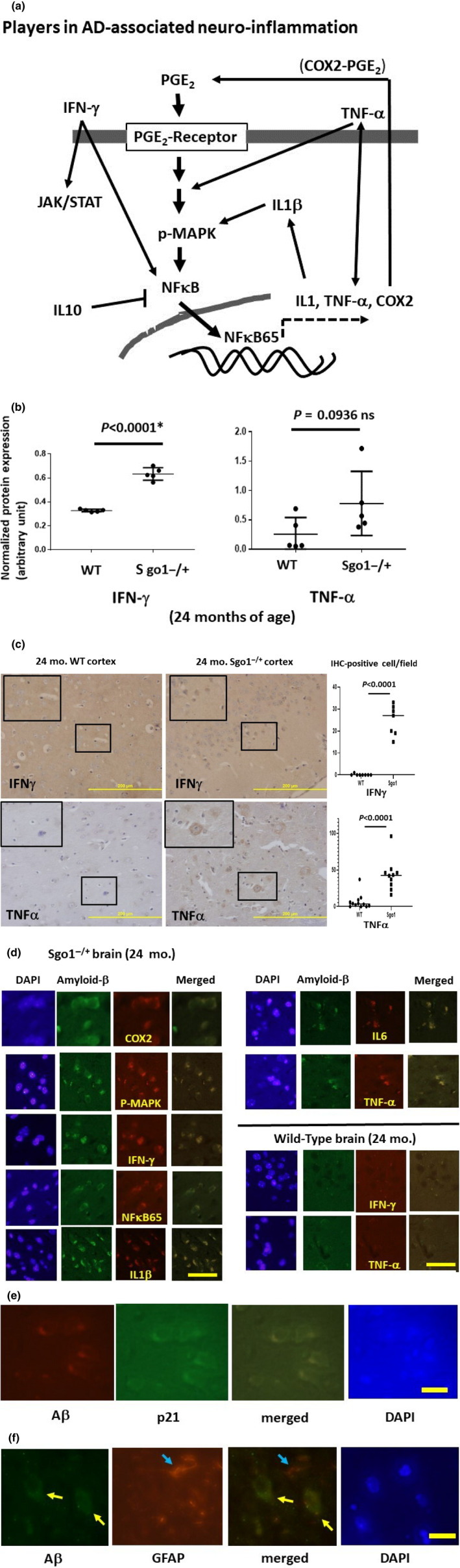
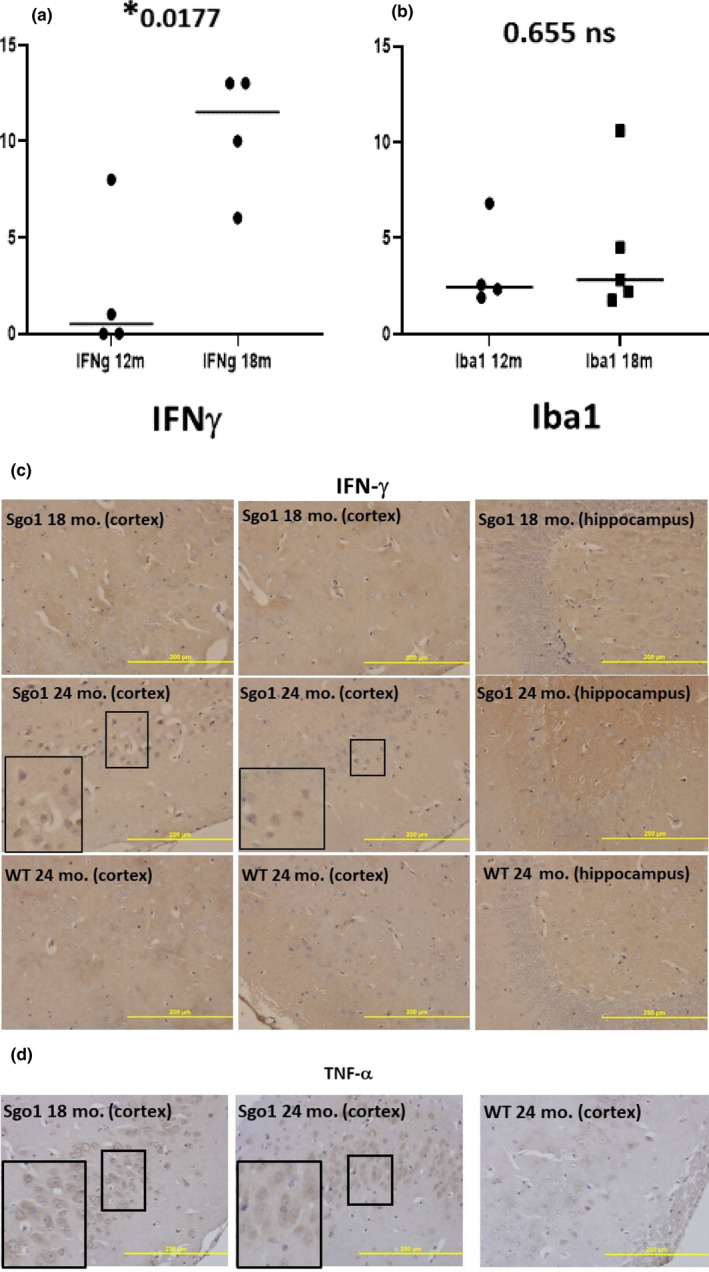
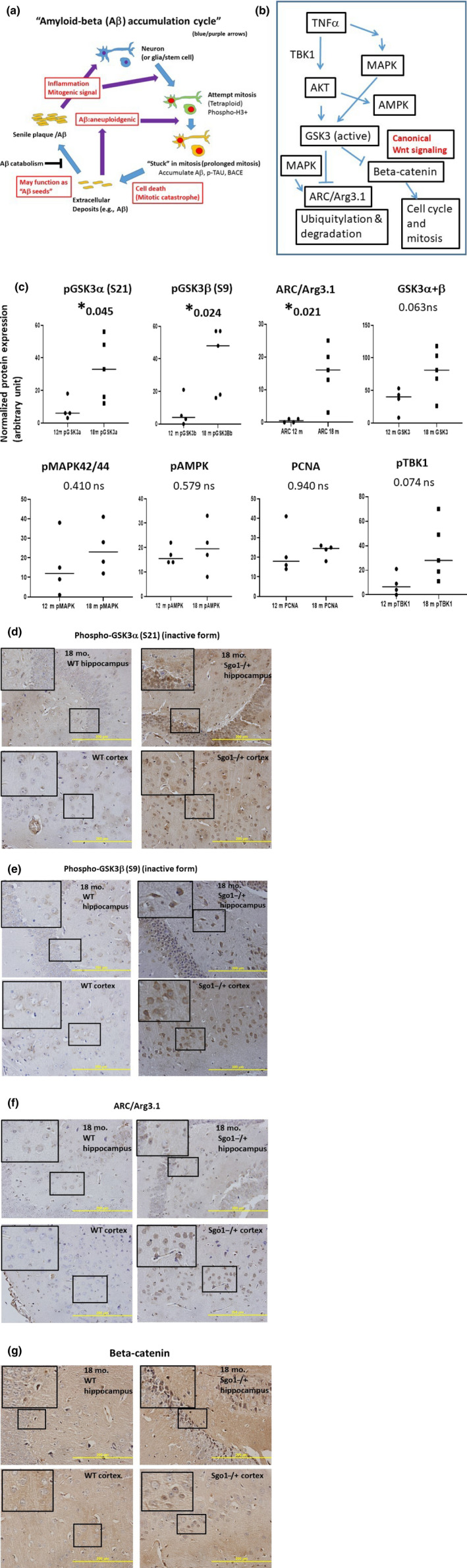
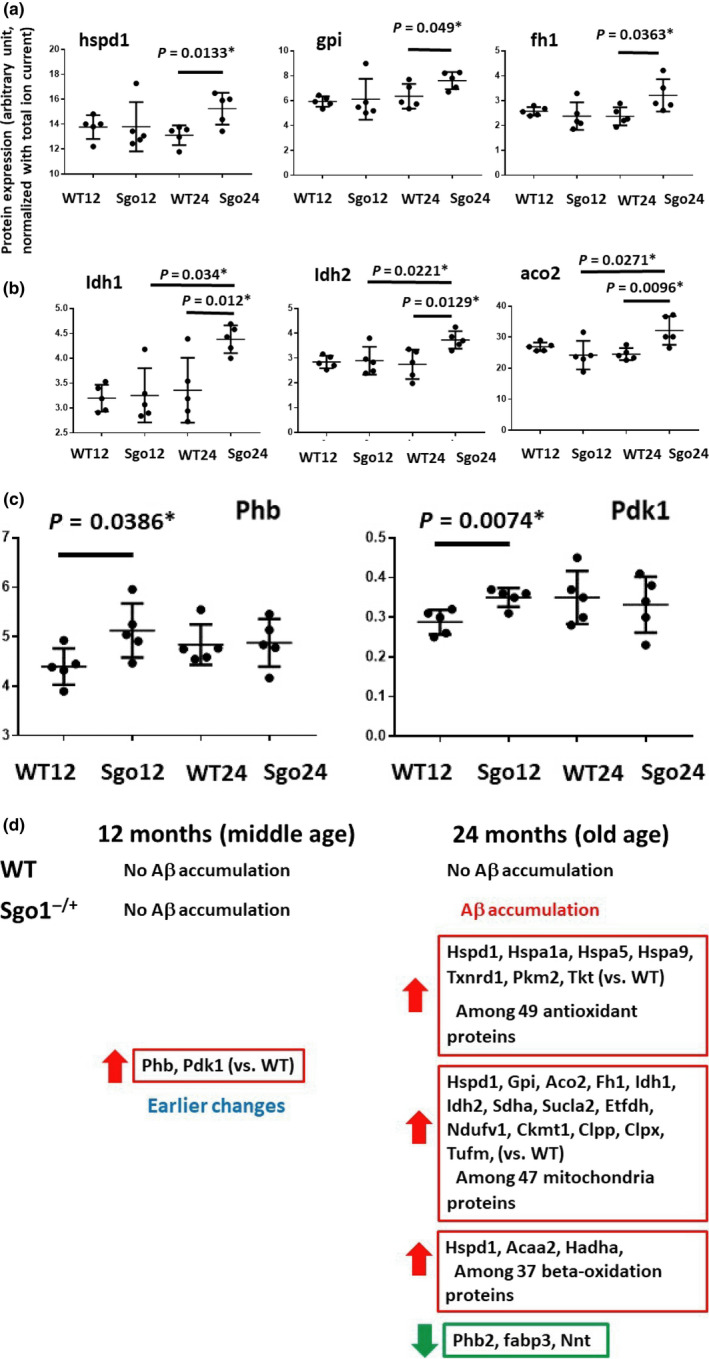
The cerebral amyloid-β accumulation that begins in middle age is considered the critical triggering event in the pathogenesis of late-onset Alzheimer's disease (LOAD). However, the molecular mechanism remains elusive. The Shugoshin 1 (Sgo1-/+ ) mouse model, a model for mitotic cohesinopathy-genomic instability that is observed in human AD at a higher rate, showed spontaneous accumulation of amyloid-β in the brain at old age. With the model, novel insights into the molecular mechanism of LOAD development are anticipated. In this study, the initial appearance of cerebral amyloid-β accumulation was determined as 15-18 months of age (late middle age) in the Sgo1-/+ model. The amyloid-β accumulation was associated with unexpected GSK3α/β inactivation, Wnt signaling activation, and ARC/Arg3.1 accumulation, suggesting involvement of both the GSK3-Arc/Arg3.1 axis and the GSK3-Wnt axis. As observed in human AD brains, neuroinflammation with IFN-γ expression occurred with amyloid-β accumulation and was pronounced in the aged (24-month-old) Sgo1-/+ model mice. AD-relevant protein panels (oxidative stress defense, mitochondrial energy metabolism, and β-oxidation and peroxisome) analysis indicated (a) early increases in Pdk1 and Phb in middle-aged Sgo1-/+ brains, and (b) misregulations in 32 proteins among 130 proteins tested in old age. Thus, initial amyloid-β accumulation in the Sgo1-/+ model is suggested to be triggered by GSK3 inactivation and the resulting Wnt activation and ARC/Arg3.1 accumulation. The model displayed characteristics and affected pathways similar to those of human LOAD including neuroinflammation, demonstrating its potential as a study tool for the LOAD development mechanism and for preclinical AD drug research and development.
Keywords: Shugoshin1; amyloid-β; cohesinopathy; genomic instability; late-onset Alzheimer's disease; mitosis; mouse model; neuroinflammation.
© 2020 The Authors. Aging Cell published by Anatomical Society and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Critical role of mitosis in spontaneous late-onset Alzheimer's disease; from a Shugoshin 1 cohesinopathy mouse model.Cell Cycle. 2018;17(19-20):2321-2334. doi: 10.1080/15384101.2018.1515554. Epub 2018 Sep 20. Cell Cycle. 2018. PMID: 30231670 Free PMC article. Review.
-
Spontaneous development of Alzheimer's disease-associated brain pathology in a Shugoshin-1 mouse cohesinopathy model.Aging Cell. 2018 Aug;17(4):e12797. doi: 10.1111/acel.12797. Epub 2018 Jun 25. Aging Cell. 2018. PMID: 29943428 Free PMC article.
-
"Amyloid-beta accumulation cycle" as a prevention and/or therapy target for Alzheimer's disease.Aging Cell. 2020 Mar;19(3):e13109. doi: 10.1111/acel.13109. Epub 2020 Jan 25. Aging Cell. 2020. PMID: 31981470 Free PMC article. Review.
-
Spatial Memory Impairment is Associated with Intraneural Amyloid-β Immunoreactivity and Dysfunctional Arc Expression in the Hippocampal-CA3 Region of a Transgenic Mouse Model of Alzheimer's Disease.J Alzheimers Dis. 2016;51(1):69-79. doi: 10.3233/JAD-150975. J Alzheimers Dis. 2016. PMID: 26836189
-
Dickkopf 3 (Dkk3) Improves Amyloid-β Pathology, Cognitive Dysfunction, and Cerebral Glucose Metabolism in a Transgenic Mouse Model of Alzheimer's Disease.J Alzheimers Dis. 2017;60(2):733-746. doi: 10.3233/JAD-161254. J Alzheimers Dis. 2017. PMID: 28922151
Cited by
-
PET molecular imaging for pathophysiological visualization in Alzheimer's disease.Eur J Nucl Med Mol Imaging. 2023 Feb;50(3):765-783. doi: 10.1007/s00259-022-05999-z. Epub 2022 Nov 14. Eur J Nucl Med Mol Imaging. 2023. PMID: 36372804 Free PMC article. Review.
-
Advance and Prospect of Positron Emission Tomography in Alzheimer's disease research.Mol Psychiatry. 2025 Jun 21. doi: 10.1038/s41380-025-03081-2. Online ahead of print. Mol Psychiatry. 2025. PMID: 40544200 Review.
-
Reducing PDK1/Akt Activity: An Effective Therapeutic Target in the Treatment of Alzheimer's Disease.Cells. 2022 May 24;11(11):1735. doi: 10.3390/cells11111735. Cells. 2022. PMID: 35681431 Free PMC article. Review.
-
GSK3α: An Important Paralog in Neurodegenerative Disorders and Cancer.Biomolecules. 2020 Dec 16;10(12):1683. doi: 10.3390/biom10121683. Biomolecules. 2020. PMID: 33339170 Free PMC article. Review.
-
Identification and Validation of Biomarkers for Alzheimer's Disease Based on Akt and Wnt Signaling Pathways in Mouse Models.Mol Neurobiol. 2025 Jul;62(7):8279-8297. doi: 10.1007/s12035-025-04785-w. Epub 2025 Feb 24. Mol Neurobiol. 2025. PMID: 39992588 Free PMC article.
References
-
- Avrahami, L. , Farfara, D. , Shaham‐Kol, M. , Vassar, R. , Frenkel, D. , & Eldar‐Finkelman, H. (2013). Inhibition of glycogen synthase kinase‐3 ameliorates β‐amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. Journal of Biological Chemistry, 288(2), 1295–1306. 10.1074/jbc.M112.409250 - DOI - PMC - PubMed
-
- Bajic, V. , Spremo‐Potparevic, B. , Zivkovic, L. , Isenovic, E. R. , & Arendt, T. (2015). Cohesion and the aneuploid phenotype in Alzheimer's disease: A tale of genome instability. Neuroscience and Biobehavioral Reviews, 55, 365–374. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
