

Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy
- PMID: 32858949
- PMCID: PMC7564605
- DOI: 10.3390/cells9091970
Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy
Abstract
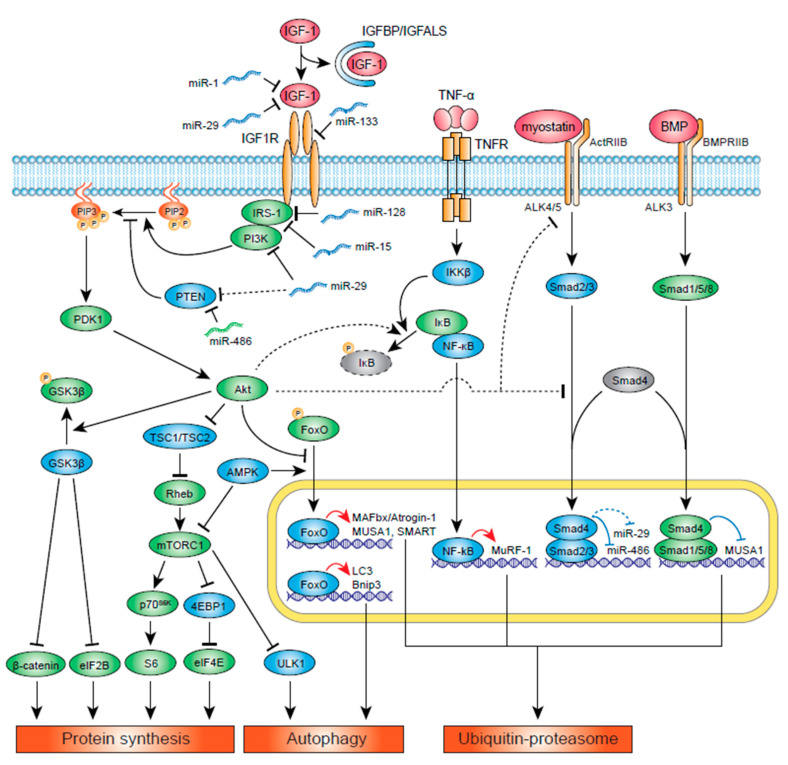
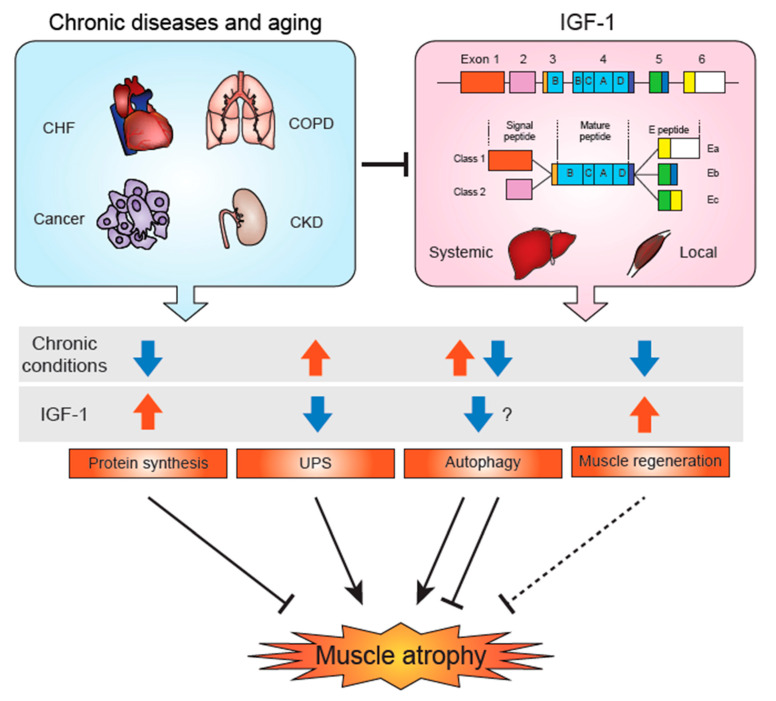
Insulin-like growth factor-1 (IGF-1) is a key growth factor that regulates both anabolic and catabolic pathways in skeletal muscle. IGF-1 increases skeletal muscle protein synthesis via PI3K/Akt/mTOR and PI3K/Akt/GSK3β pathways. PI3K/Akt can also inhibit FoxOs and suppress transcription of E3 ubiquitin ligases that regulate ubiquitin proteasome system (UPS)-mediated protein degradation. Autophagy is likely inhibited by IGF-1 via mTOR and FoxO signaling, although the contribution of autophagy regulation in IGF-1-mediated inhibition of skeletal muscle atrophy remains to be determined. Evidence has suggested that IGF-1/Akt can inhibit muscle atrophy-inducing cytokine and myostatin signaling via inhibition of the NF-κΒ and Smad pathways, respectively. Several miRNAs have been found to regulate IGF-1 signaling in skeletal muscle, and these miRs are likely regulated in different pathological conditions and contribute to the development of muscle atrophy. IGF-1 also potentiates skeletal muscle regeneration via activation of skeletal muscle stem (satellite) cells, which may contribute to muscle hypertrophy and/or inhibit atrophy. Importantly, IGF-1 levels and IGF-1R downstream signaling are suppressed in many chronic disease conditions and likely result in muscle atrophy via the combined effects of altered protein synthesis, UPS activity, autophagy, and muscle regeneration.
Keywords: atrophy; autophagy; cachexia; hypertrophy; insulin-like growth factor-1; muscle regeneration; skeletal muscle.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Majorczyk M., Smolag D. Effect of physical activity on IGF-1 and IGFBP levels in the context of civilization diseases prevention. Rocz. Panstw. Zakl. Hig. 2016;67:105–111. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
