

Recent developments in diagnostics and treatment of neonatal cholestasis
- PMID: 32861449
- PMCID: PMC7459146
- DOI: 10.1016/j.sempedsurg.2020.150945
Recent developments in diagnostics and treatment of neonatal cholestasis
Abstract
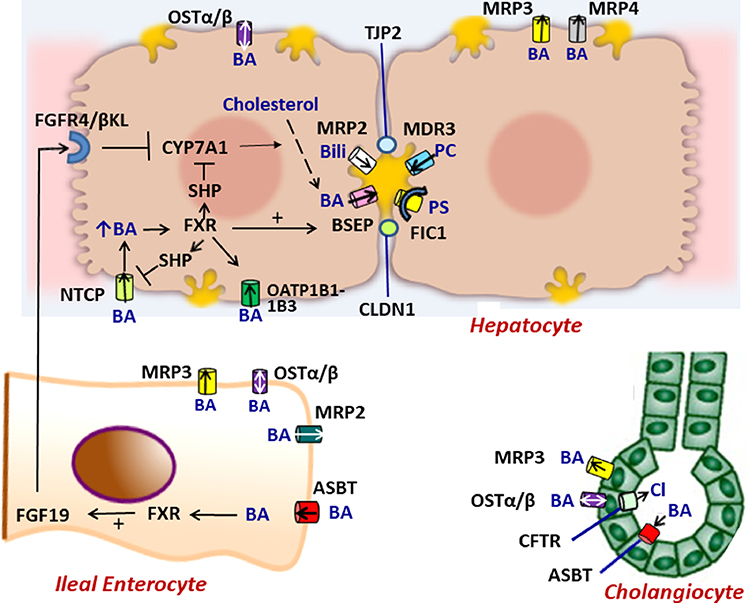
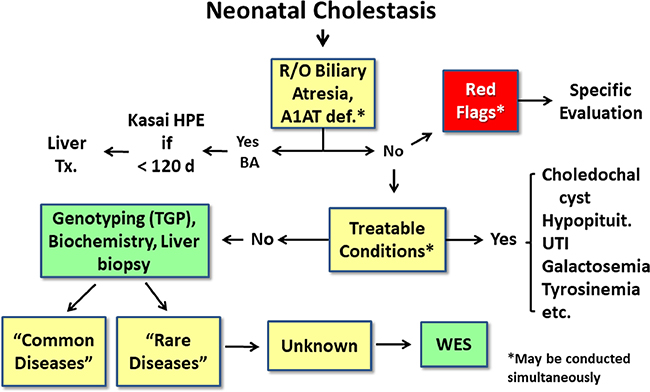
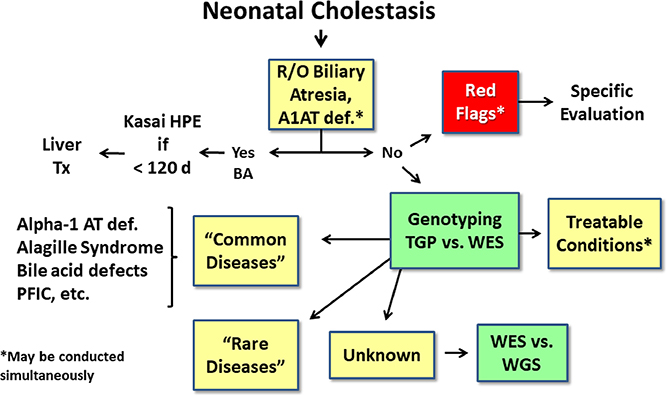
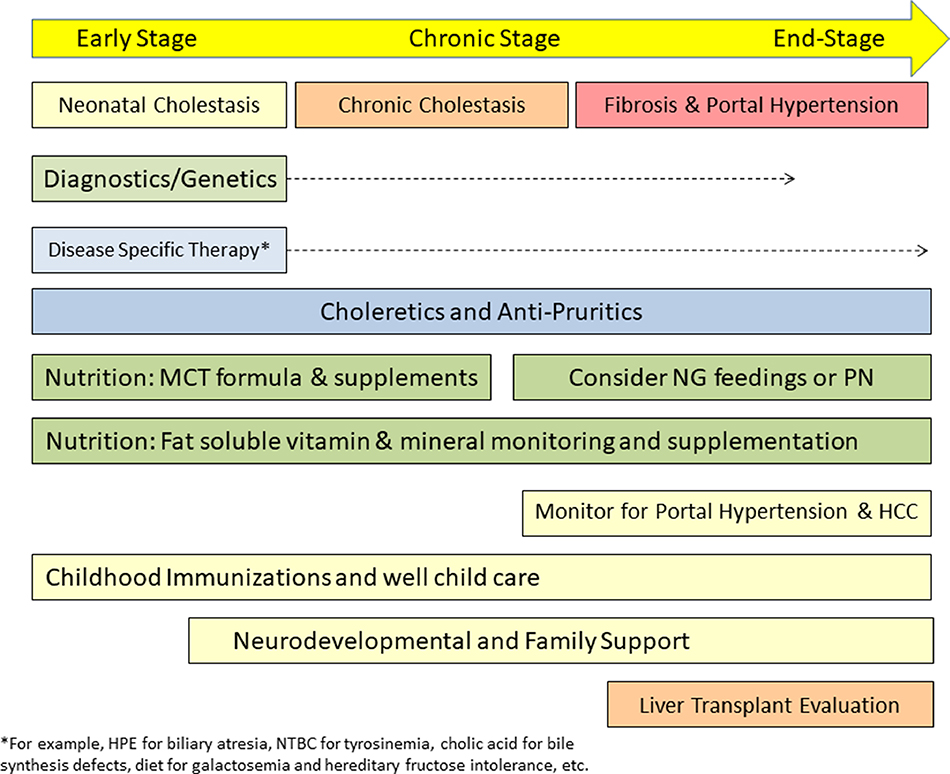
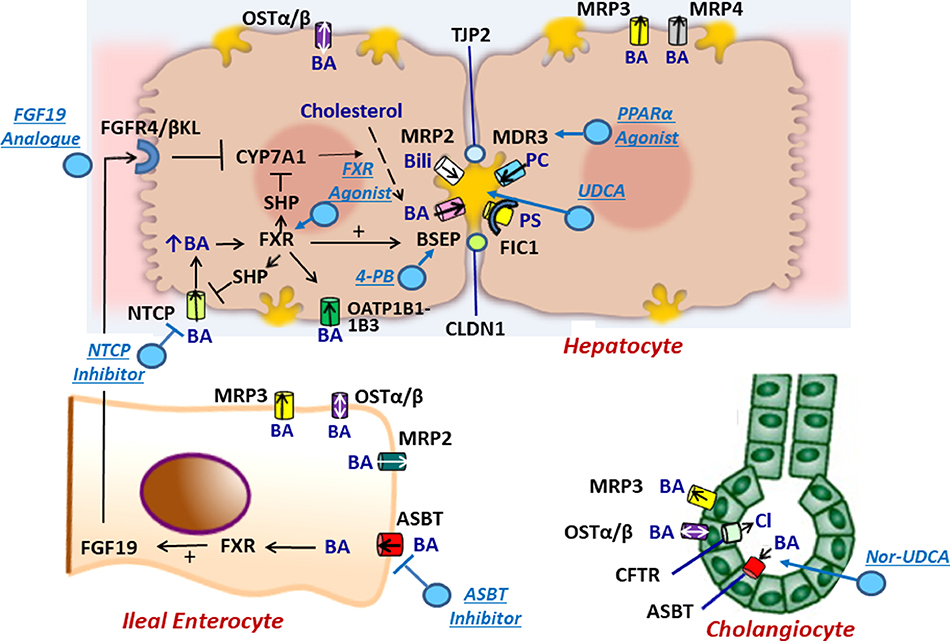
Neonatal cholestasis is characterized by conjugated hyperbilirubinemia in the newborn and young infant and is a sign common to over 100 hepatobiliary and/or metabolic disorders. A timely evaluation for its etiology is critical in order to quickly identify treatable causes such as biliary atresia, many of which benefit from early therapy. An expanding group of molecularly defined disorders involving bile formation, canalicular transporters, tight junction proteins and inborn errors of metabolism are being continuously discovered because of advances in genetic testing and bioinformatics. The advent of next generation sequencing has transformed our ability to test for multiple genes and whole exome or whole genome sequencing within days to weeks, enabling rapid and affordable molecular diagnosis for disorders that cannot be directly diagnosed from standard blood tests or liver biopsy. Thus, our diagnostic algorithms for neonatal cholestasis are undergoing transformation, moving genetic sequencing to earlier in the evaluation pathway once biliary atresia, "red flag" disorders and treatable disorders are excluded. Current therapies focus on promoting bile flow, reducing pruritus, ensuring optimal nutrition, and monitoring for complications, without addressing the underlying cause of cholestasis in most instances. Our improved understanding of bile formation and the enterohepatic circulation of bile acids has led to emerging therapies for cholestasis which require appropriate pediatric clinical trials. Despite these advances, the cause and optimal therapy for biliary atresia remain elusive. The goals of this review are to outline the etiologies, diagnostic pathways and current and emerging management strategies for neonatal cholestasis.
Keywords: Alagille syndrome; Biliary atresia; Genomics; Neonatal cholestasis; Progressive familial intrahepatic cholestasis.
Copyright © 2020. Published by Elsevier Inc.
Conflict of interest statement
Declaration of Competing Interest None.
Figures
References
-
- Feldman A, Suchy FJ. Approach to the infant with cholestasis In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver Disease in Children New York City: Cambridge University Press; 2014, p. 101–10.
-
- Harpavat S, Garcia-Prats JA, Shneider BL. Newborn Bilirubin Screening for Biliary Atresia. N Engl J Med 2016;375(6):605–6. - PubMed
-
- Harpavat S, Ramraj R, Finegold MJ, Brandt ML, Hertel PM, Fallon SC, et al. Newborn Direct or Conjugated Bilirubin Measurements As a Potential Screen for Biliary Atresia. J Pediatr Gastroenterol Nutr 2016;62(6):799–803. - PubMed
-
- Chardot C, Buet C, Serinet MO, Golmard JL, Lachaux A, Roquelaure B, et al. Improving outcomes of biliary atresia: French national series 1986–2009. Journal of hepatology 2013;58(6):1209–17. - PubMed
