

Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction
- PMID: 32863203
- PMCID: PMC7381685
- DOI: 10.1016/j.redox.2020.101638
Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction
Erratum in
-
Erratum to Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction, Redox Biology 36 (2020) 101638.Redox Biol. 2021 Dec;48:102200. doi: 10.1016/j.redox.2021.102200. Epub 2021 Nov 27. Redox Biol. 2021. PMID: 34844898 Free PMC article. No abstract available.
Abstract
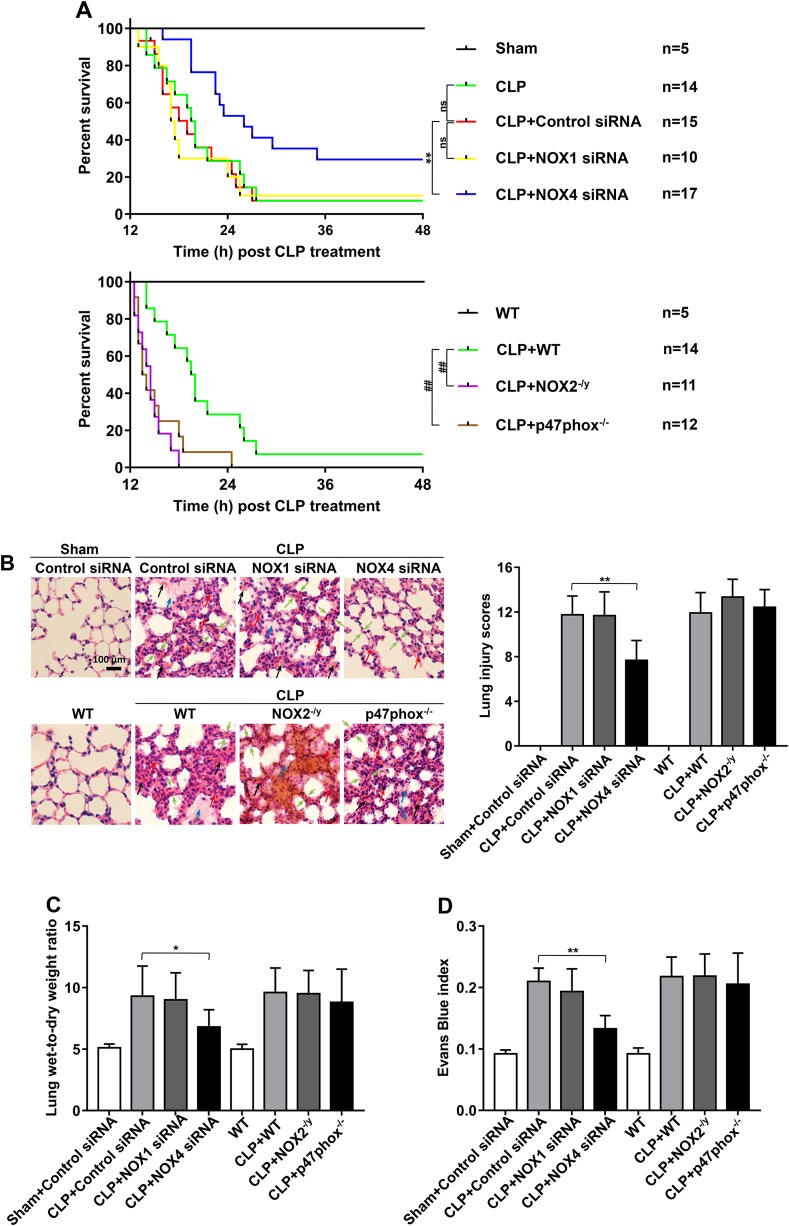
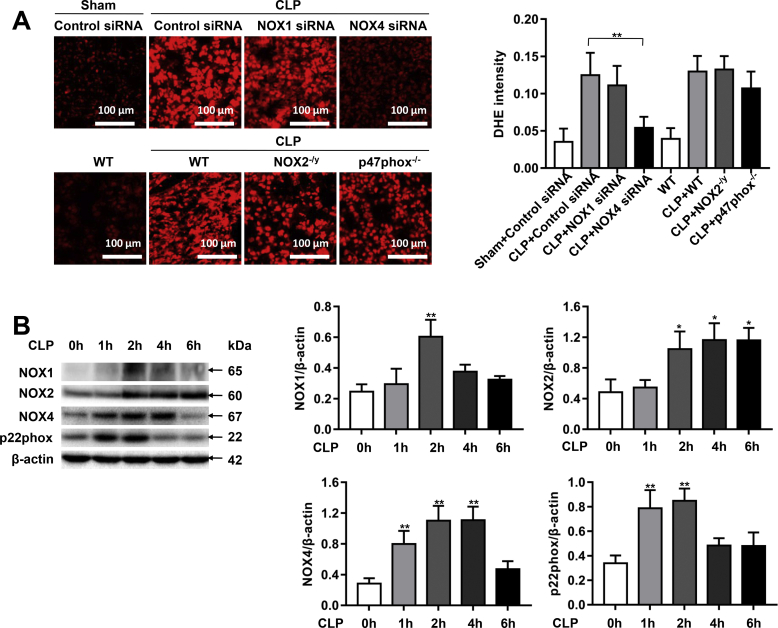
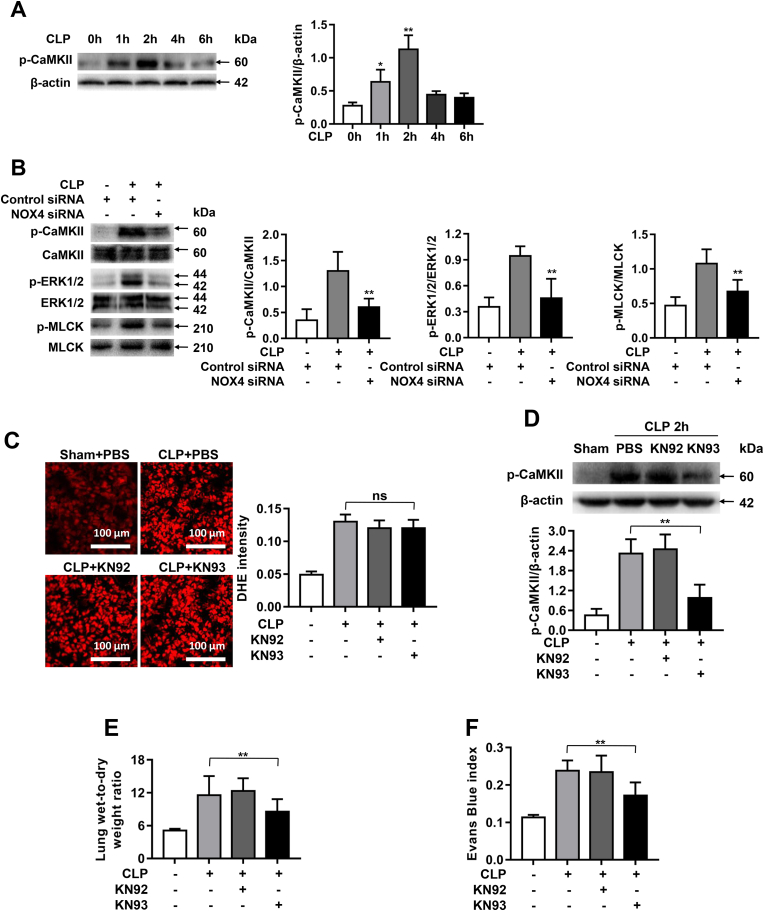
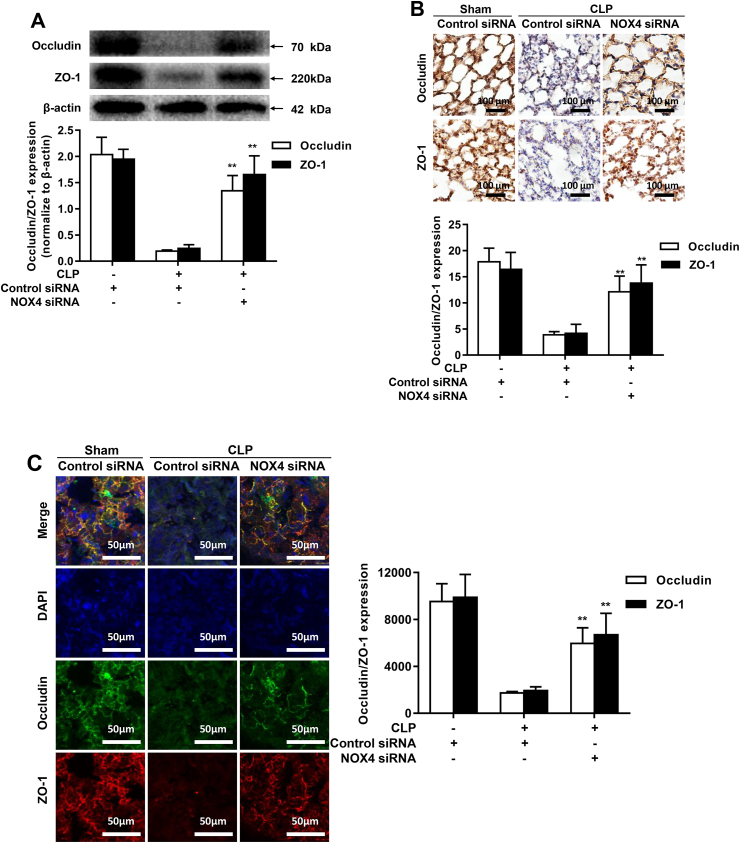
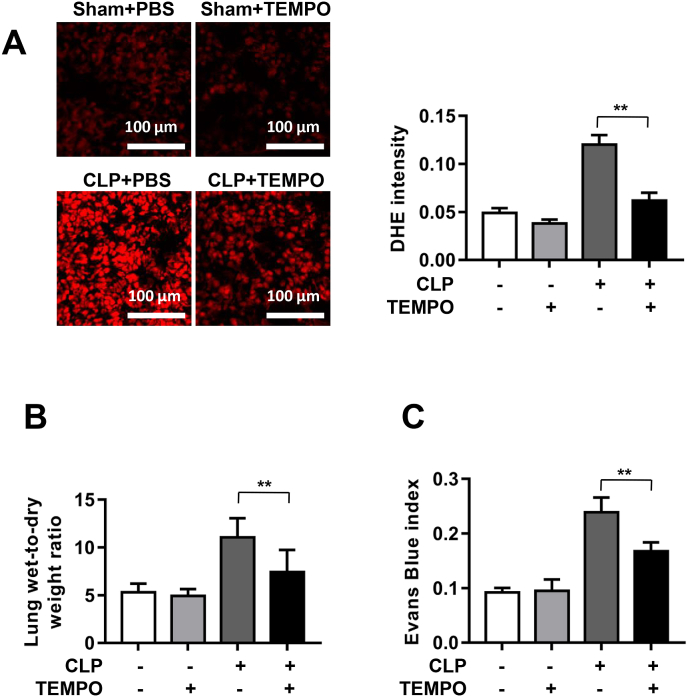
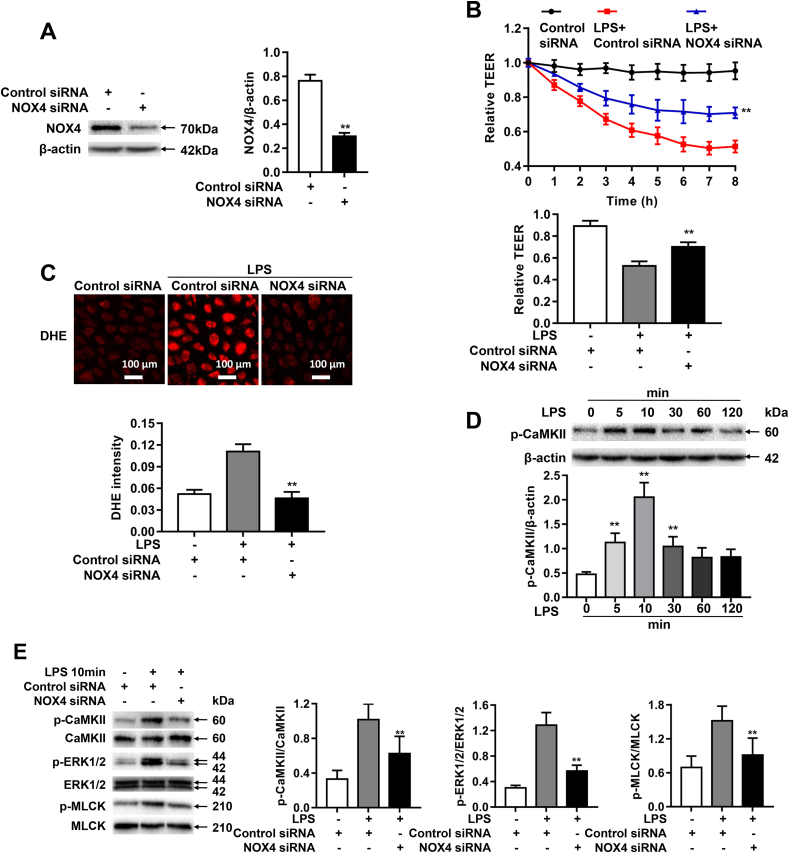
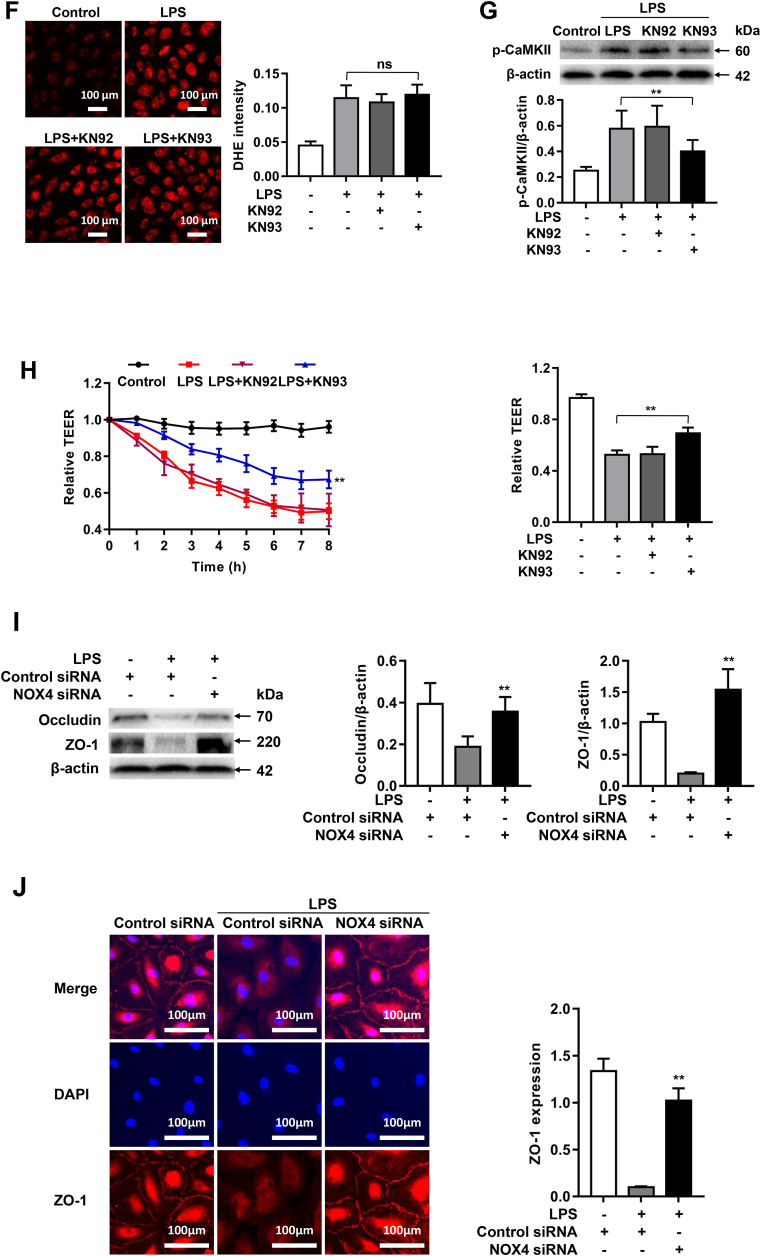
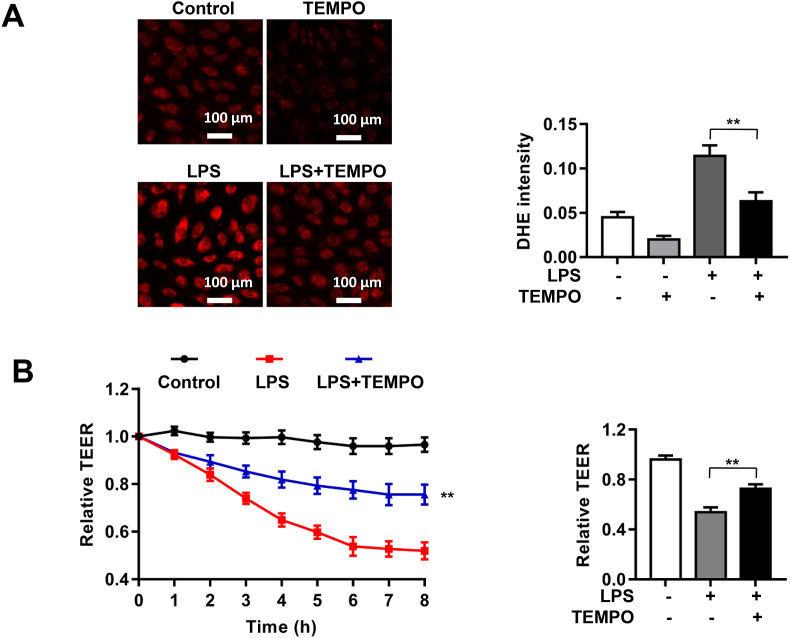
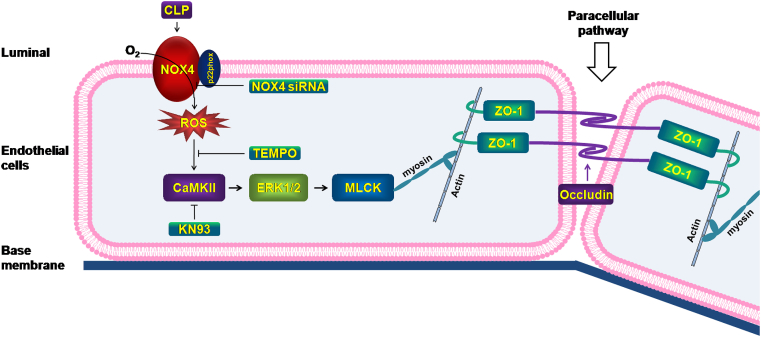
Increased pulmonary vascular permeability due to endothelial cell (EC) barrier dysfunction is a major pathological feature of acute respiratory distress syndrome/acute lung injury (ARDS/ALI), which is a devastating critical illness with high incidence and excessive mortality. Activation of NADPH oxidase (NOX) induces EC dysfunction via production of reactive oxygen species (ROS). However, the role(s) of NOX isoform(s), and their downstream signaling events, in the development of ARDS/ALI have remained unclear. Cecal Ligation Puncture (CLP) was used to induce preclinical septic ALI in wild-type mice and mice deficient in NOX2 or p47phox, or mice transfected of control siRNA, NOX1 or NOX4 siRNA in vivo. The survival rate of the CLP group at 24 h (26.6%, control siRNA treated) was substantially improved by NOX4 knockdown (52.9%). Mice lacking NOX2 or p47phox, however, had worse outcomes after CLP (survival rates at 0% and 8.3% respectively), whereas NOX1-silenced mice had similar survival rate (30%). NOX4 knockdown attenuated lung ROS production in septic mice, whereas NOX1 knockdown, NOX2 knockout, or p47phox knockout in mice had no effects. In addition, NOX4 knockdown attenuated redox-sensitive activation of the CaMKII/ERK1/2/MLCK pathway, and restored expression of EC tight junction proteins ZO-1 and Occludin to maintain EC barrier integrity. Correspondingly, NOX4 knockdown in cultured human lung microvascular ECs also reduced LPS-induced ROS production, CaMKII/ERK1/2/MLCK activation and EC barrier dysfunction. Scavenging superoxide in vitro and in vivo with TEMPO, or inhibiting CaMKII activation with KN93, had similar effects as NOX4 knockdown in preserving EC barrier dysfunction. In summary, we have identified a novel, selective and causal role of NOX4 (versus other NOX isoforms) in inducing lung EC barrier dysfunction and injury/mortality in a preclinical CLP-induced septic model, which involves redox-sensitive activation of CaMKII/ERK1/2/MLCK pathway. Targeting NOX4 may therefore prove to an innovative therapeutic option that is markedly effective in treating ALI/ARDS.
Keywords: Acute lung injury (ALI); Acute respiratory distress syndrome (ARDS); Endothelial barrier dysfunction; Endothelial cell (EC); Endothelial permeability; NADPH oxidase (NOX); NOX1; NOX2; NOX4; Occludin; Reactive oxygen species (ROS); Tight junction; ZO-1; p22phox; p47phox.
Copyright © 2020 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Fan E., Brodie D., Slutsky A.S. Acute respiratory distress syndrome: advances in diagnosis and treatment. JAMA. 2018;319:698–710. - PubMed
-
- Bellani G., Laffey J.G., Pham T., Fan E., Brochard L., Esteban A., Gattinoni L., van Haren F., Larsson A., McAuley D.F., Ranieri M., Rubenfeld G., Thompson B.T., Wrigge H., Slutsky A.S., Pesenti A. Epidemiology, patterns of Care, and mortality for patients with acute respiratory distress syndrome in intensive Care units in 50 countries. JAMA. 2016;315:788–800. - PubMed
-
- Dudek S.M., Garcia J.G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. (Bethesda, Md. 1985) 2001;91:1487–1500. - PubMed
-
- McVerry B.J., Garcia J.G. Endothelial cell barrier regulation by sphingosine 1-phosphate. J. Cell. Biochem. 2004;92:1075–1085. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
