

Comprehensive evolution and molecular characteristics of a large number of SARS-CoV-2 genomes reveal its epidemic trends
- PMID: 32866640
- PMCID: PMC7455167
- DOI: 10.1016/j.ijid.2020.08.066
Comprehensive evolution and molecular characteristics of a large number of SARS-CoV-2 genomes reveal its epidemic trends
Abstract
Objectives: To further reveal the phylogenetic evolution and molecular characteristics of the whole genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) based on a large number of genomes and provide a basis for the prevention and treatment of SARS-CoV-2.
Methods: Various evolution analysis methods were employed.
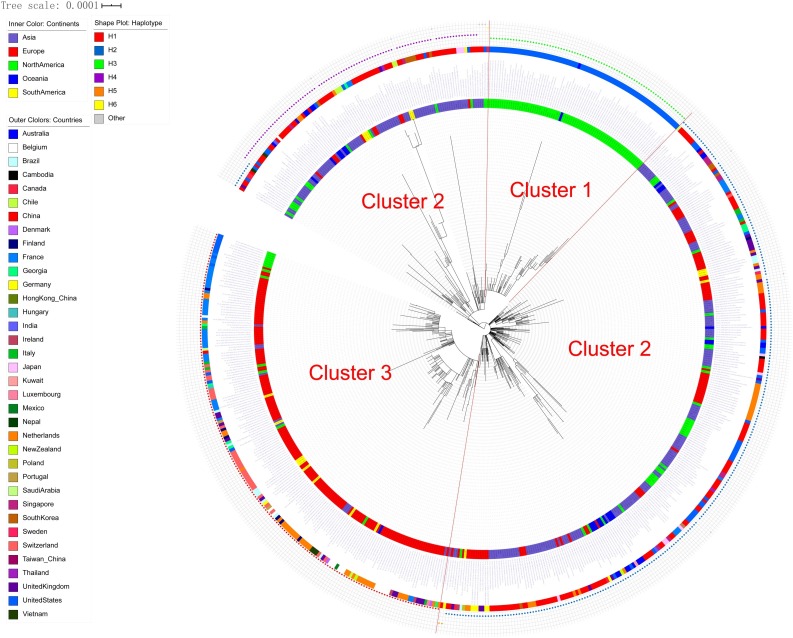
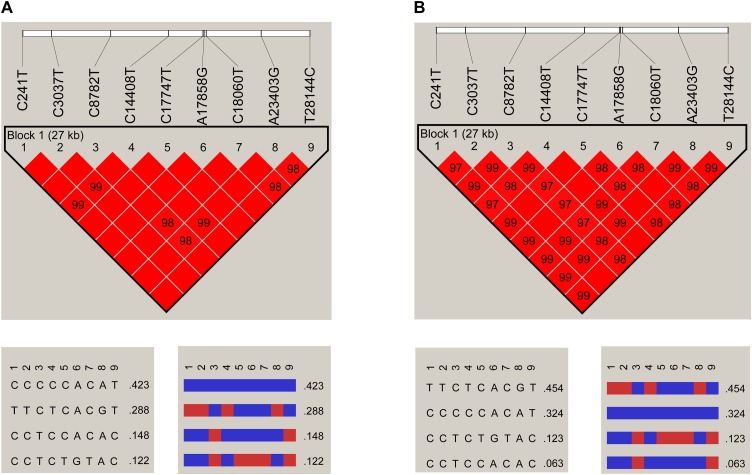
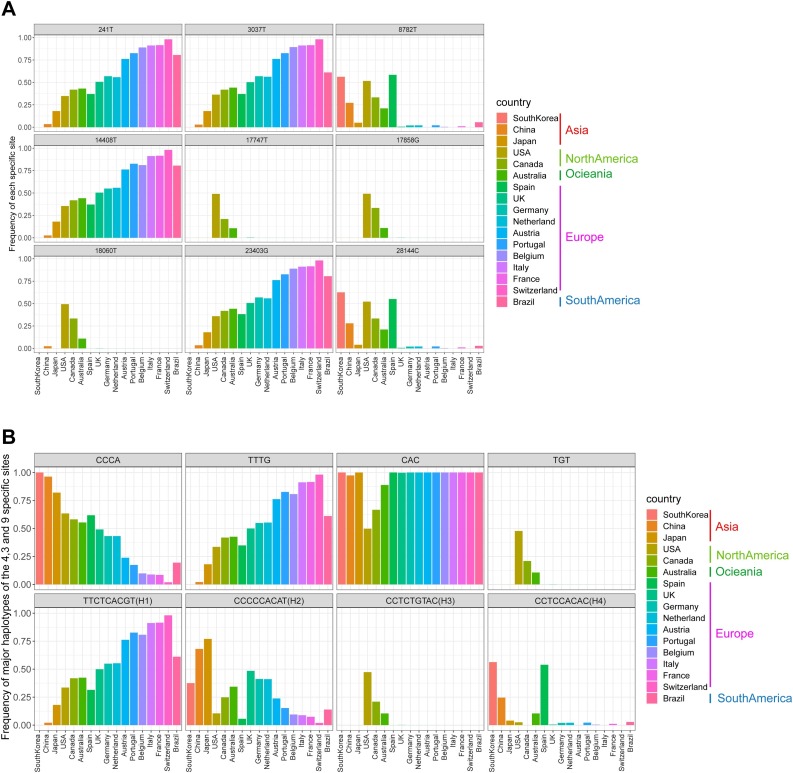
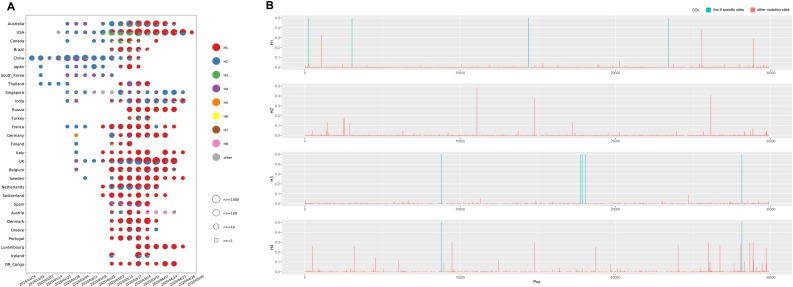
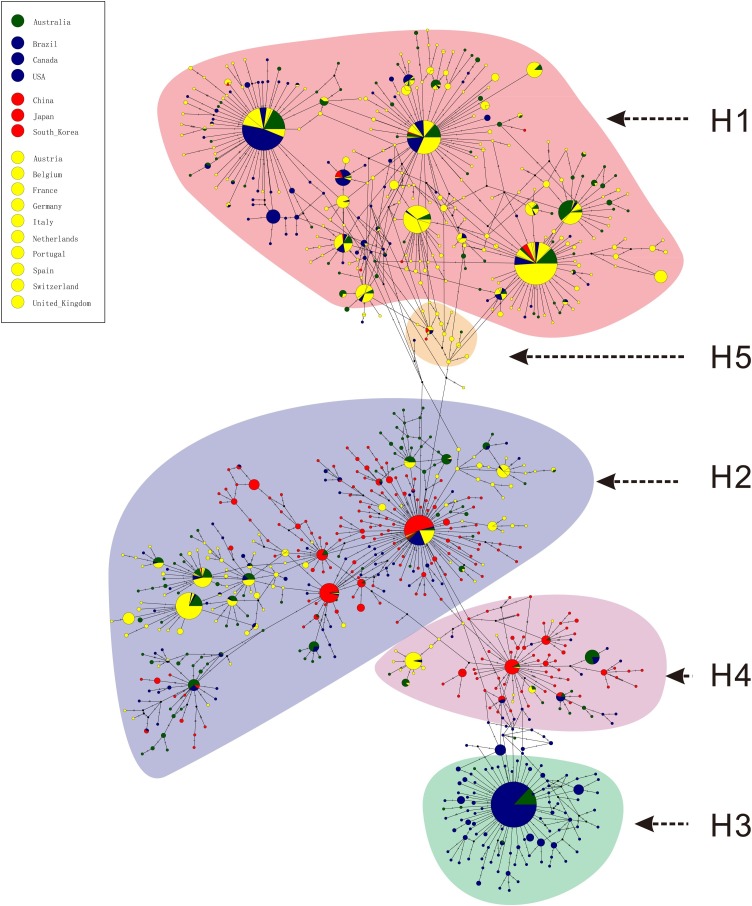
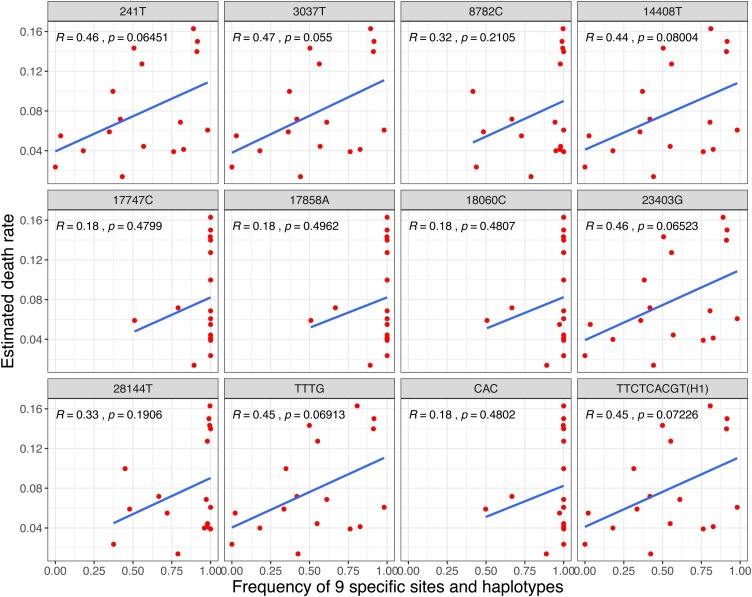
Results: The estimated ratio of the rates of non-synonymous to synonymous changes (Ka/Ks) of SARS-CoV-2 was 1.008 or 1.094 based on 622 or 3624 SARS-CoV-2 genomes and nine key specific sites of high linkage, and four major haplotypes were found: H1, H2, H3 and H4. The results of Ka/Ks, detected population size and development trends of each major haplotype showed that H3 and H4 subgroups were going through a purify evolution and almost disappeared after detection, indicating that they might have existed for a long time. The H1 and H2 subgroups were going through a near neutral or neutral evolution and globally increased with time, and the frequency of H1 was generally high in Europe and correlated with the death rate (r >0.37), suggesting that these two haplotypes might relate to the infectivity or pathogenicity of SARS-CoV-2.
Conclusions: Several key specific sites and haplotypes related to the infectivity or pathogenicity of SARS-CoV-2, and the possible earlier origin time and place of SARS-CoV-2 were indicated based on the evolution and epidemiology of 16,373 SARS-CoV-2 genomes.
Keywords: Classification; Evolution; Haplotype; SARS-CoV-2.
Copyright © 2020 The Authors. Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors report no declarations of interest.
Figures
References
-
- Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
