

A very early diagnosis of Alstrӧm syndrome by next generation sequencing
- PMID: 32867697
- PMCID: PMC7460749
- DOI: 10.1186/s12881-020-01110-1
A very early diagnosis of Alstrӧm syndrome by next generation sequencing
Abstract
Background: Alström syndrome is a rare recessively inherited disorder caused by variants in the ALMS1 gene. It is characterized by multiple organ dysfunction, including cone-rod retinal dystrophy, dilated cardiomyopathy, hearing loss, obesity, insulin resistance, hyperinsulinemia, type 2 diabetes mellitus and systemic fibrosis. Heterogeneity and age-dependent development of clinical manifestations make it difficult to obtain a clear diagnosis, especially in pediatric patients.
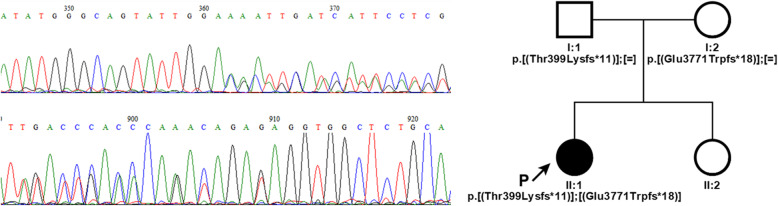
Case presentation: Here we report the case of a girl with Alström syndrome. Genetic examination was proposed at age 22 months when suspected macular degeneration was the only major finding. Next generation sequencing of a panel of genes linked to eye-related pathologies revealed two compound heterozygous variants in the ALMS1 gene. Frameshift variants c.1196_1202del, p.(Thr399Lysfs*11), rs761292021 and c.11310_11313del, (p.Glu3771Trpfs*18), rs747272625 were detected in exons 5 and 16, respectively. Both variants cause frameshifts and generation of a premature stop-codon that probably leads to mRNA nonsense-mediated decay. Validation and segregation of ALMS1 variants were confirmed by Sanger sequencing.
Conclusions: Genetic testing makes it possible, even in childhood, to increase the number of correct diagnoses of patients who have ambiguous phenotypes caused by rare genetic variants. The development of high-throughput sequencing technologies offers an exceptionally valuable screening tool for clear genetic diagnoses and ensures early multidisciplinary management and treatment of the emerging symptoms.
Keywords: ALMS1; Alström syndrome; Case report; Next generation sequencing.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
Similar articles
-
Identification of novel compound heterozygous variants of the ALMS1 gene in a child with Alström syndrome by whole genome sequencing.Gene. 2024 Dec 15;929:148827. doi: 10.1016/j.gene.2024.148827. Epub 2024 Aug 8. Gene. 2024. PMID: 39122231
-
Novel Mutations of the ALMS1 Gene in Patients with Alström Syndrome.Intern Med. 2021 Dec 1;60(23):3721-3728. doi: 10.2169/internalmedicine.6467-20. Epub 2021 Jun 19. Intern Med. 2021. PMID: 34148947 Free PMC article.
-
A Next Generation Sequencing custom gene panel as first line diagnostic tool for atypical cases of syndromic obesity: Application in a case of Alström syndrome.Eur J Med Genet. 2018 Feb;61(2):79-83. doi: 10.1016/j.ejmg.2017.10.016. Epub 2017 Oct 24. Eur J Med Genet. 2018. PMID: 29079548
-
A novel variant in ALMS1 in a patient with Alström syndrome and prenatal diagnosis for the fetus in the family: A case report and literature review.Mol Med Rep. 2020 Oct;22(4):3271-3276. doi: 10.3892/mmr.2020.11398. Epub 2020 Jul 31. Mol Med Rep. 2020. PMID: 32945434 Free PMC article. Review.
-
The phenotypic and molecular genetic spectrum of Alström syndrome in 44 Turkish kindreds and a literature review of Alström syndrome in Turkey.J Hum Genet. 2015 Jan;60(1):1-9. doi: 10.1038/jhg.2014.85. Epub 2014 Oct 9. J Hum Genet. 2015. PMID: 25296579 Free PMC article. Review.
Cited by
-
Identification of a Rare Exon 19 Skipping Mutation in ALMS1 Gene in Alström Syndrome Patients From Two Unrelated Saudi Families.Front Pediatr. 2021 Apr 26;9:652011. doi: 10.3389/fped.2021.652011. eCollection 2021. Front Pediatr. 2021. PMID: 33981653 Free PMC article.
-
Next-Generation Sequencing of a Large Gene Panel for Outcome Prediction of Bariatric Surgery in Patients with Severe Obesity.J Clin Med. 2022 Dec 19;11(24):7531. doi: 10.3390/jcm11247531. J Clin Med. 2022. PMID: 36556146 Free PMC article.
-
Genotype-phenotype associations in Alström syndrome: a systematic review and meta-analysis.J Med Genet. 2023 Dec 21;61(1):18-26. doi: 10.1136/jmg-2023-109175. J Med Genet. 2023. PMID: 37321834 Free PMC article.
-
New pathogenic variants of ALMS1 gene in two Chinese families with Alström Syndrome.BMC Ophthalmol. 2022 Sep 26;22(1):386. doi: 10.1186/s12886-022-02597-3. BMC Ophthalmol. 2022. PMID: 36162988 Free PMC article.
References
-
- Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31:74–78. - PubMed
-
- Marshall JD, Beck S, Maffei P, Naggert JK. Alström syndrome. Eur J Hum Genet. 2007;15:1193–1202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
