

Benzo(a)pyrene exposure induced neuronal loss, plaque deposition, and cognitive decline in APP/PS1 mice
- PMID: 32867800
- PMCID: PMC7461337
- DOI: 10.1186/s12974-020-01925-y
Benzo(a)pyrene exposure induced neuronal loss, plaque deposition, and cognitive decline in APP/PS1 mice
Abstract
Background: Exposure to benzo(a)pyrene (BaP) was associated with cognitive impairments and some Alzheimer's disease (AD)-like pathological changes. However, it is largely unknown whether BaP exposure participates in the disease progression of AD.
Objectives: To investigate the effect of BaP exposure on AD progression and its underlying mechanisms.
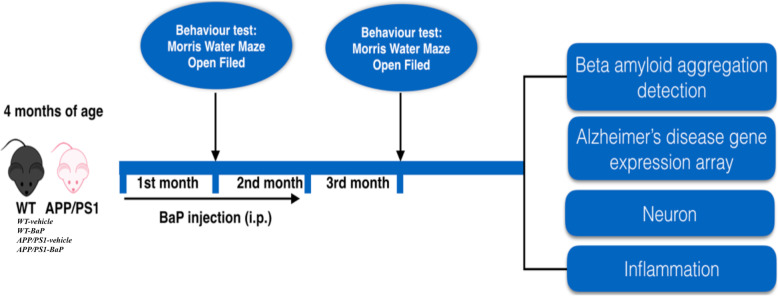
Methods: BaP or vehicle was administered to 4-month-old APPswe/PS1dE9 transgenic (APP/PS1) mice and wildtype (WT) mice for 2 months. Learning and memory ability and exploratory behaviors were evaluated 1 month after the initiation/termination of BaP exposure. AD-like pathological and biochemical alterations were examined 1 month after 2-month BaP exposure. Levels of soluble beta-amyloid (Aβ) oligomers and the number of Aβ plaques in the cortex and the hippocampus were quantified. Gene expression profiling was used to evaluate alternation of genes/pathways associated with AD onset and progression. Immunohistochemistry and Western blot were used to demonstrate neuronal loss and neuroinflammation in the cortex and the hippocampus. Treatment of primary neuron-glia cultures with aged Aβ (a mixture of monomers, oligomers, and fibrils) and/or BaP was used to investigate mechanisms by which BaP enhanced Aβ-induced neurodegeneration.
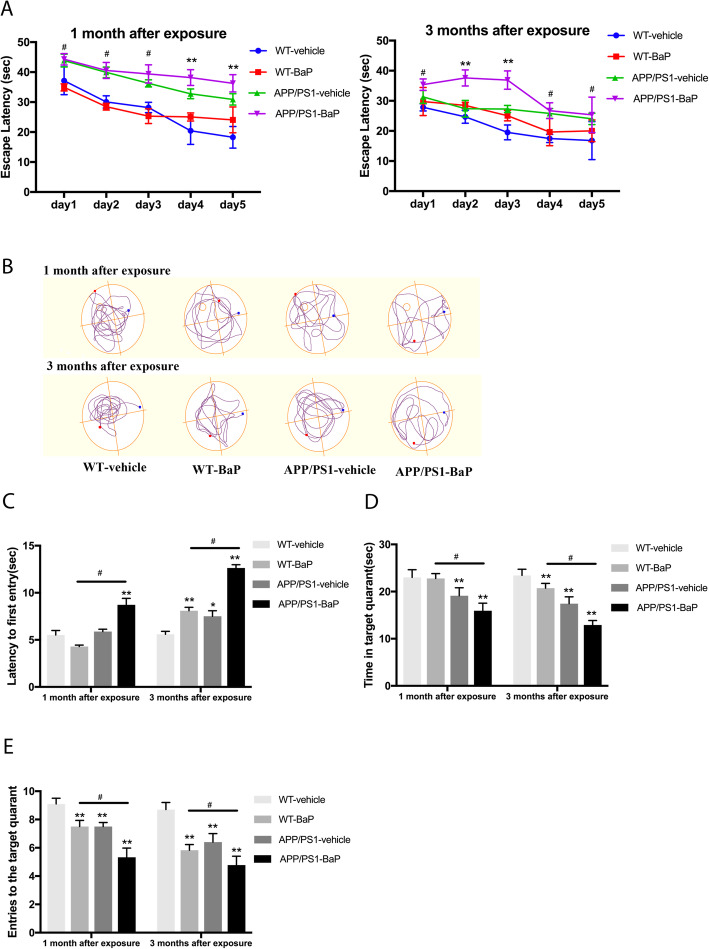
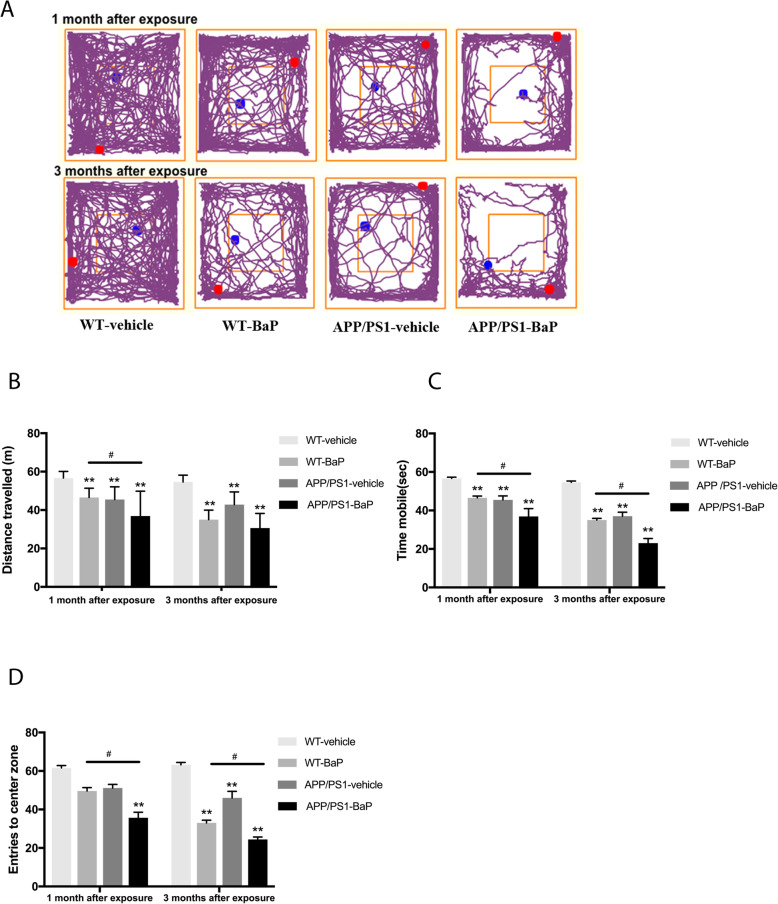
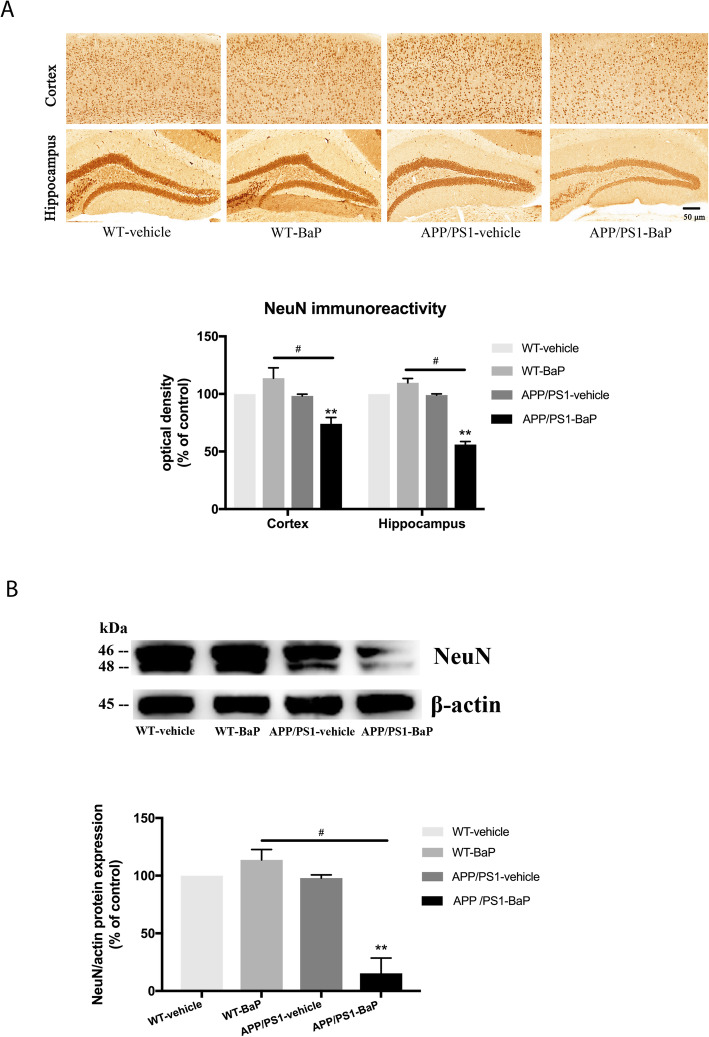
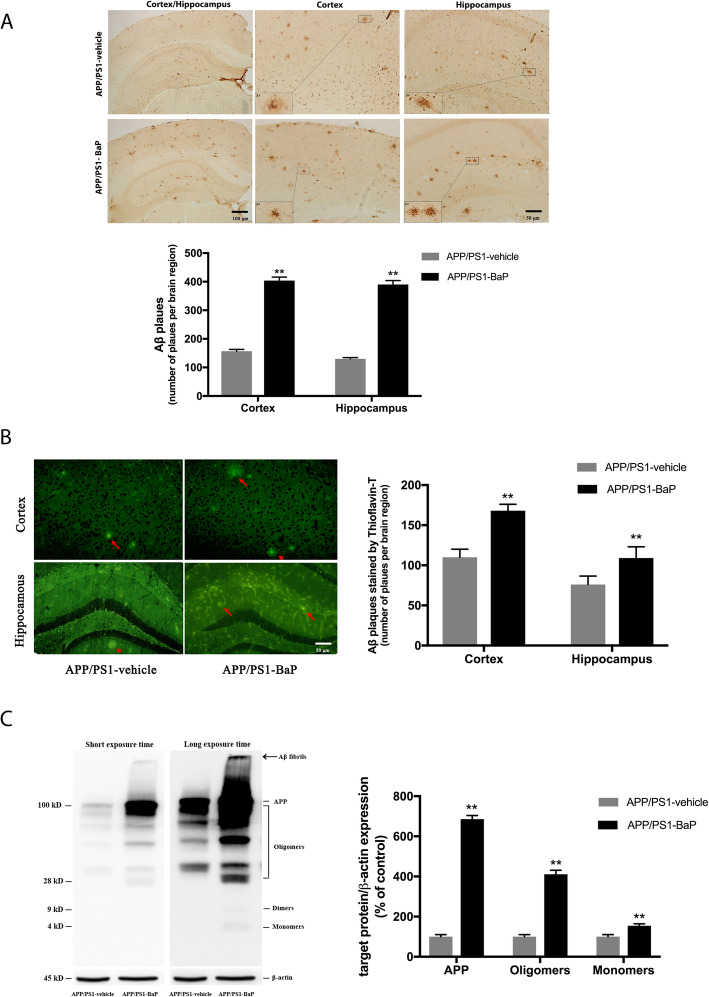
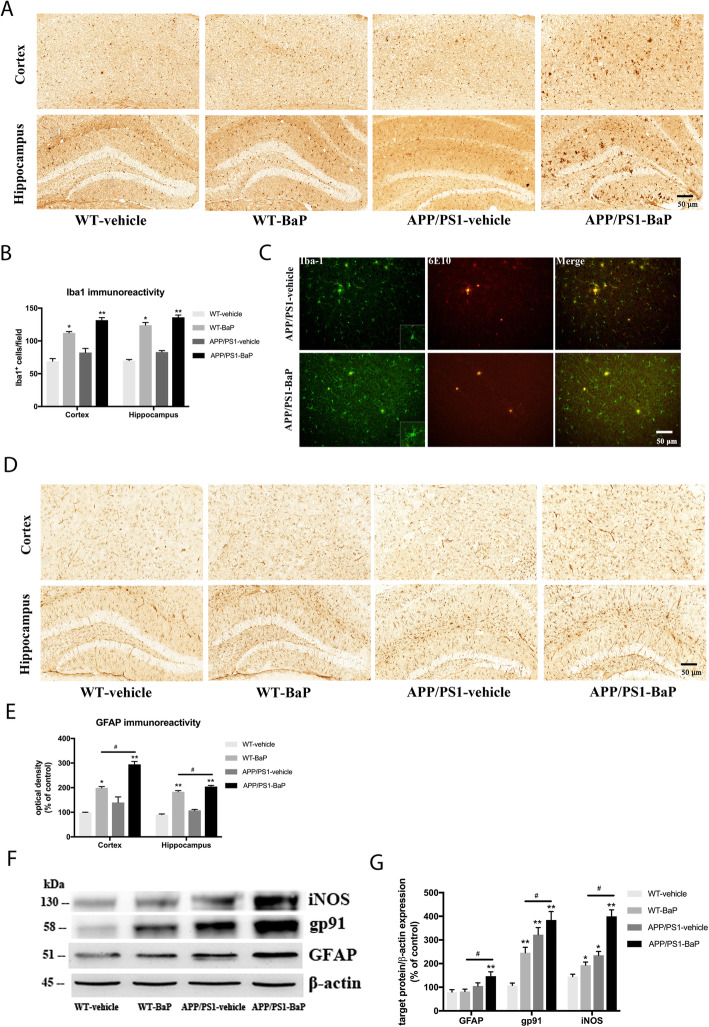
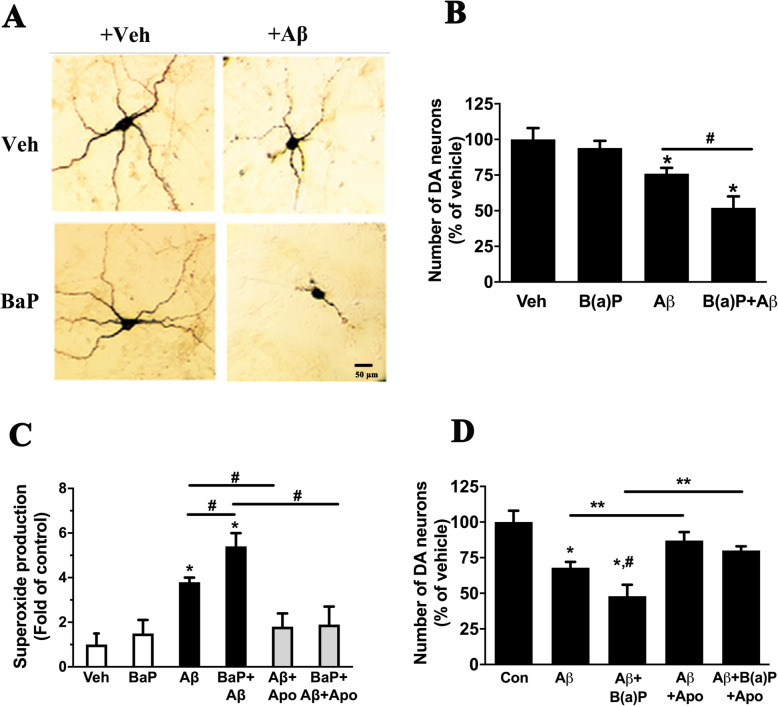
Results: BaP exposure induced progressive decline in spatial learning/memory and exploratory behaviors in APP/PS1 mice and WT mice, and APP/PS1 mice showed severer behavioral deficits than WT mice. Moreover, BaP exposure promoted neuronal loss, Aβ burden and Aβ plaque formation in APP/PS1 mice, but not in WT mice. Gene expression profiling showed most robust alteration in genes and pathways related to inflammation and immunoregulatory process, Aβ secretion and degradation, and synaptic formation in WT and APP/PS1 mice after BaP exposure. Consistently, the cortex and the hippocampus of WT and APP/PS1 mice displayed activation of microglia and astroglia and upregulation of inducible nitric oxide synthase (iNOS), glial fibrillary acidic protein (GFAP), and NADPH oxidase (three widely used neuroinflammatory markers) after BaP exposure. Furthermore, BaP exposure aggravated neurodegeneration induced by aged Aβ peptide in primary neuron-glia cultures through enhancing NADPH oxidase-derived oxidative stress.
Conclusion: Our study showed that chronic exposure to environmental pollutant BaP induced, accelerated, and exacerbated the progression of AD, in which elevated neuroinflammation and NADPH oxidase-derived oxidative insults were key pathogenic events.
Keywords: Alzheimer’s disease; Amyloid; Benzo(a)pyrene; Cognition; Neuroinflammation.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet. 2016;388:505–517. - PubMed
-
- Calderon-Garciduenas L, Reed W, Maronpot RR, Henriquez-Roldan C, Delgado-Chavez R, Calderon-Garciduenas A, et al. Brain inflammation and Alzheimer's-like pathology in individuals exposed to severe air pollution. Toxicol Pathol. 2004;32:650–658. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
