

Geldanamycin-Derived HSP90 Inhibitors Are Synthetic Lethal with NRF2
- PMID: 32868290
- PMCID: PMC7588872
- DOI: 10.1128/MCB.00377-20
Geldanamycin-Derived HSP90 Inhibitors Are Synthetic Lethal with NRF2
Abstract
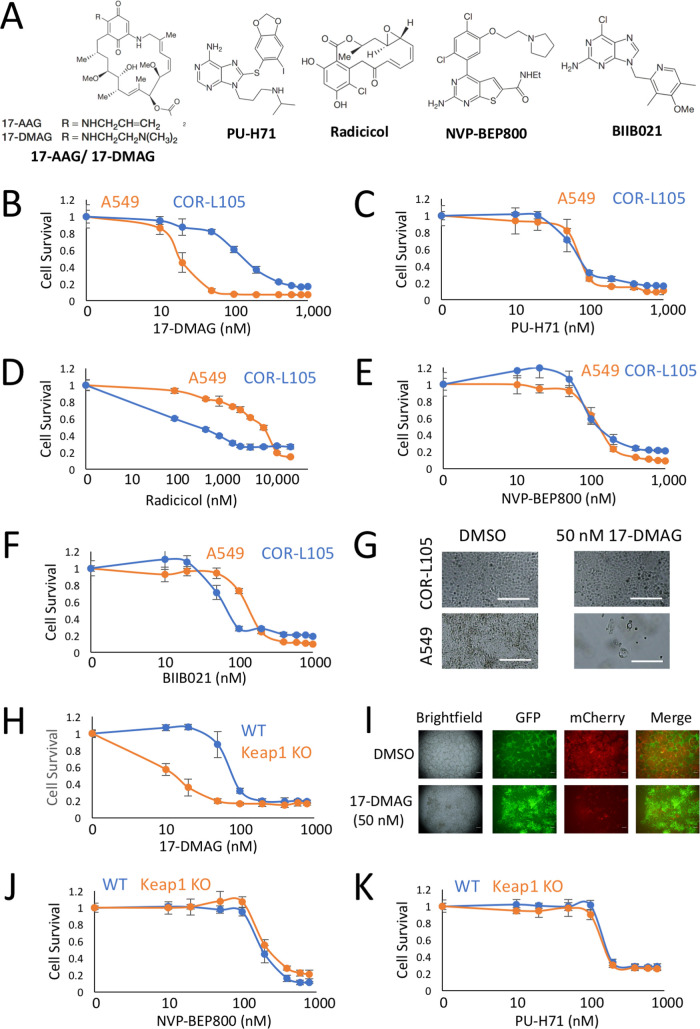
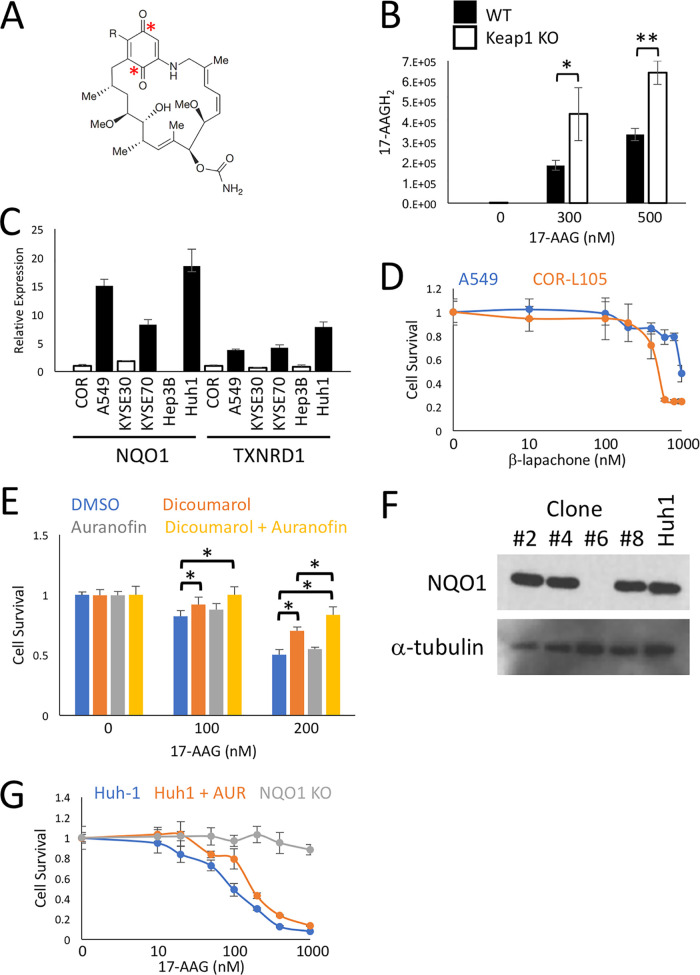
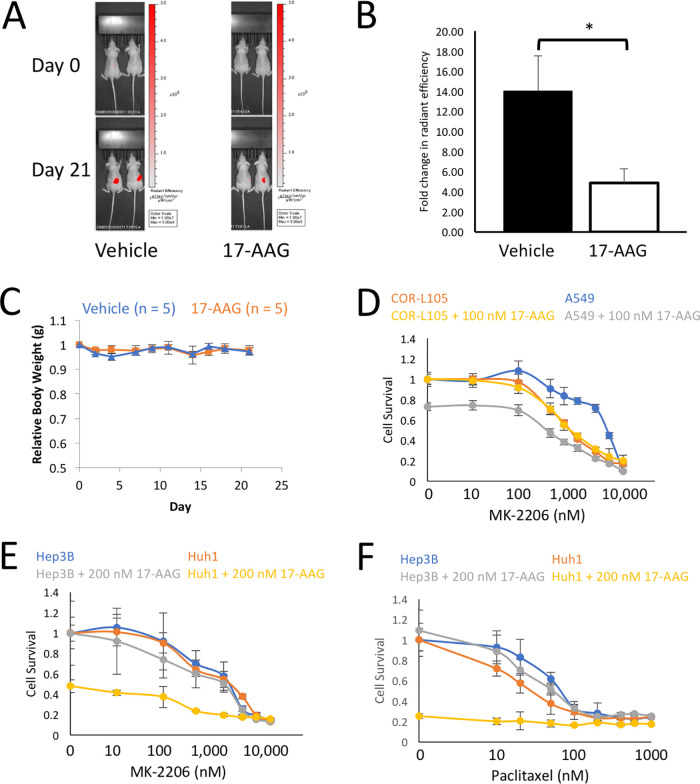
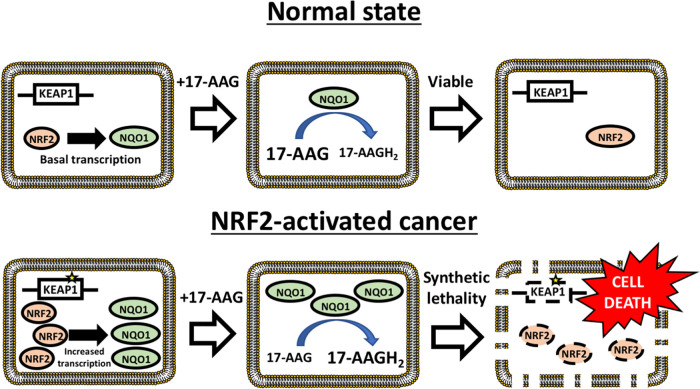
Activating mutations in KEAP1-NRF2 are frequently found in tumors of the lung, esophagus, and liver, where they are associated with aggressive growth, resistance to cancer therapies, and low overall survival. Despite the fact that NRF2 is a validated driver of tumorigenesis and chemotherapeutic resistance, there are currently no approved drugs which can inhibit its activity. Therefore, there is an urgent clinical need to identify NRF2-selective cancer therapies. To this end, we developed a novel synthetic lethal assay, based on fluorescently labeled isogenic wild-type and Keap1 knockout cell lines, in order to screen for compounds which selectively kill cells in an NRF2-dependent manner. Through this approach, we identified three compounds based on the geldanamycin scaffold which display synthetic lethality with NRF2. Mechanistically, we show that products of NRF2 target genes metabolize the quinone-containing geldanamycin compounds into more potent HSP90 inhibitors, which enhances their cytotoxicity while simultaneously restricting the synthetic lethal effect to cells with aberrant NRF2 activity. As all three of the geldanamycin-derived compounds have been used in clinical trials, they represent ideal candidates for drug repositioning to target the currently untreatable NRF2 activity in cancer.
Keywords: KEAP1; NRF2; Nfe2l2; cancer; oxidative stress; synthetic lethal.
Copyright © 2020 American Society for Microbiology.
Figures





References
-
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, Bailey MH, Liang WW, Foltz SM, Shmulevich I, Ding L, Heins Z, Ochoa A, Gross B, Gao J, Zhang H, Kundra R, Kandoth C, Bahceci I, Dervishi L, Dogrusoz U, Zhou W, Shen H, Laird PW, Way GP, Greene CS, Liang H, Xiao Y, Wang C, Iavarone A, Berger AH, Bivona TG, Lazar AJ, Hammer GD, Giordano T, Kwong LN, McArthur G, Huang C, Tward AD, Frederick MJ, McCormick F, Meyerson M, Van Allen EM, Cancer Genome Atlas Research Network, et al. 2018. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173:321–337.e10. doi:10.1016/j.cell.2018.03.035. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials