

Massively parallel and time-resolved RNA sequencing in single cells with scNT-seq
- PMID: 32868927
- PMCID: PMC8103797
- DOI: 10.1038/s41592-020-0935-4
Massively parallel and time-resolved RNA sequencing in single cells with scNT-seq
Abstract
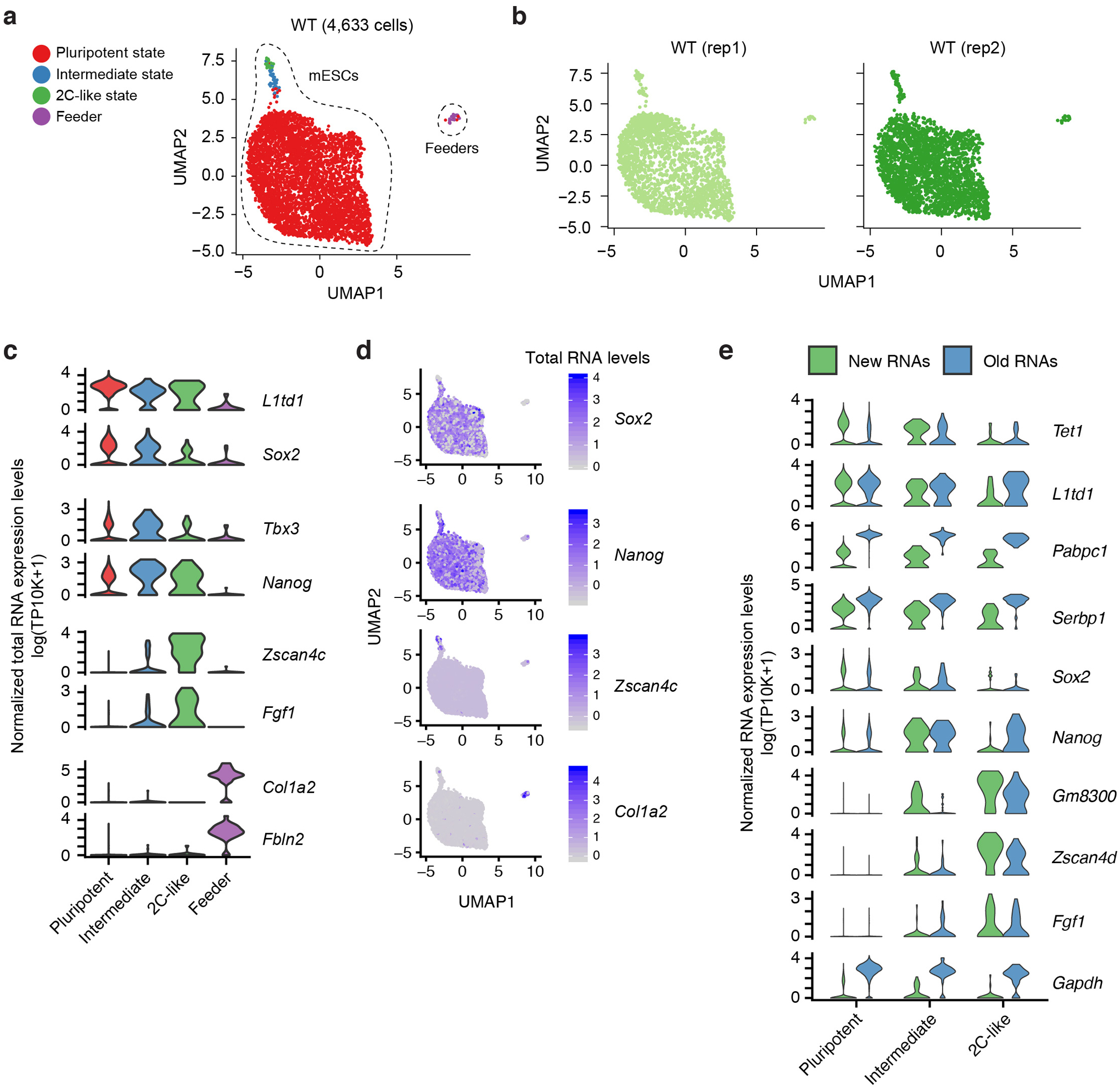
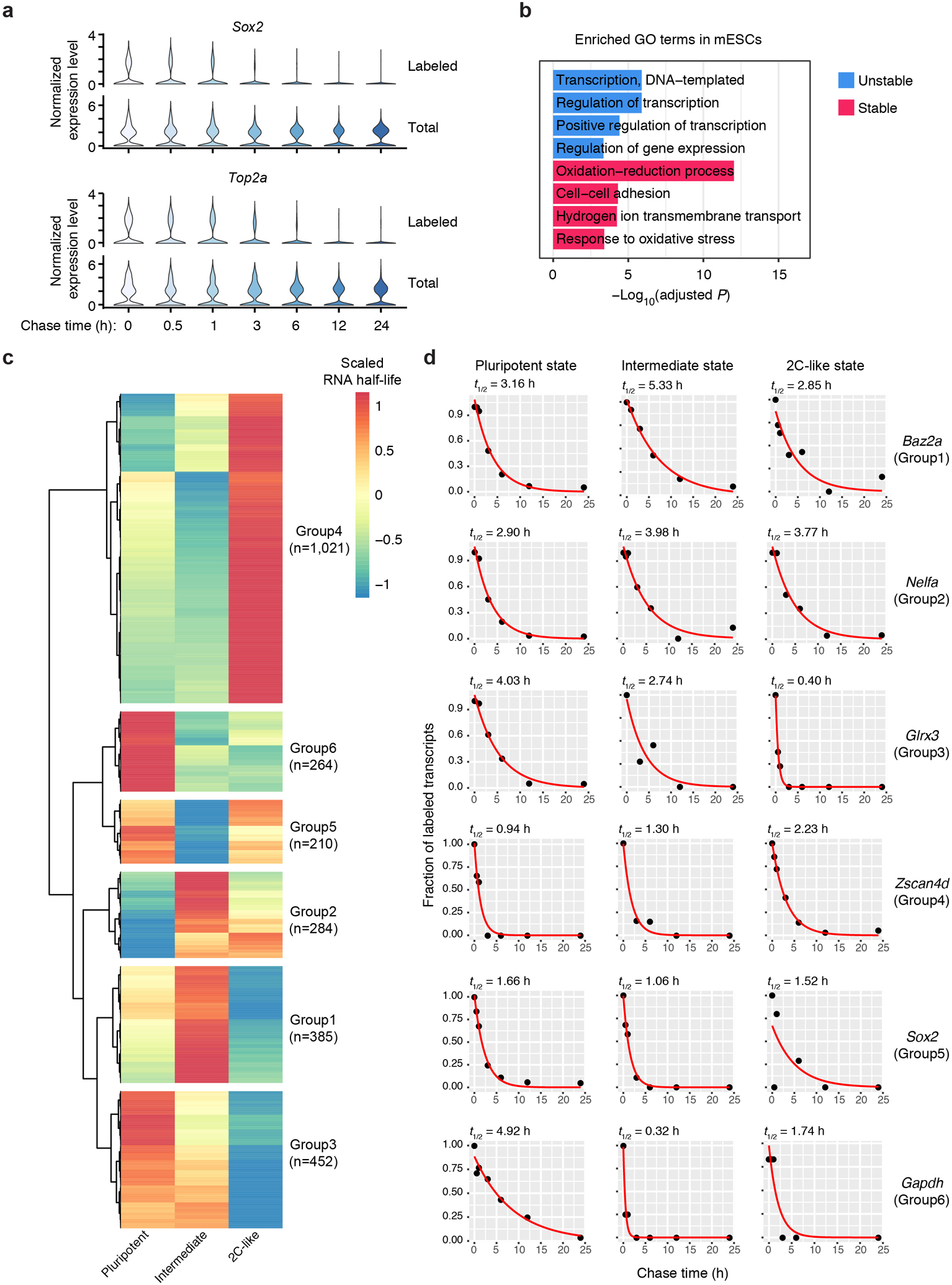
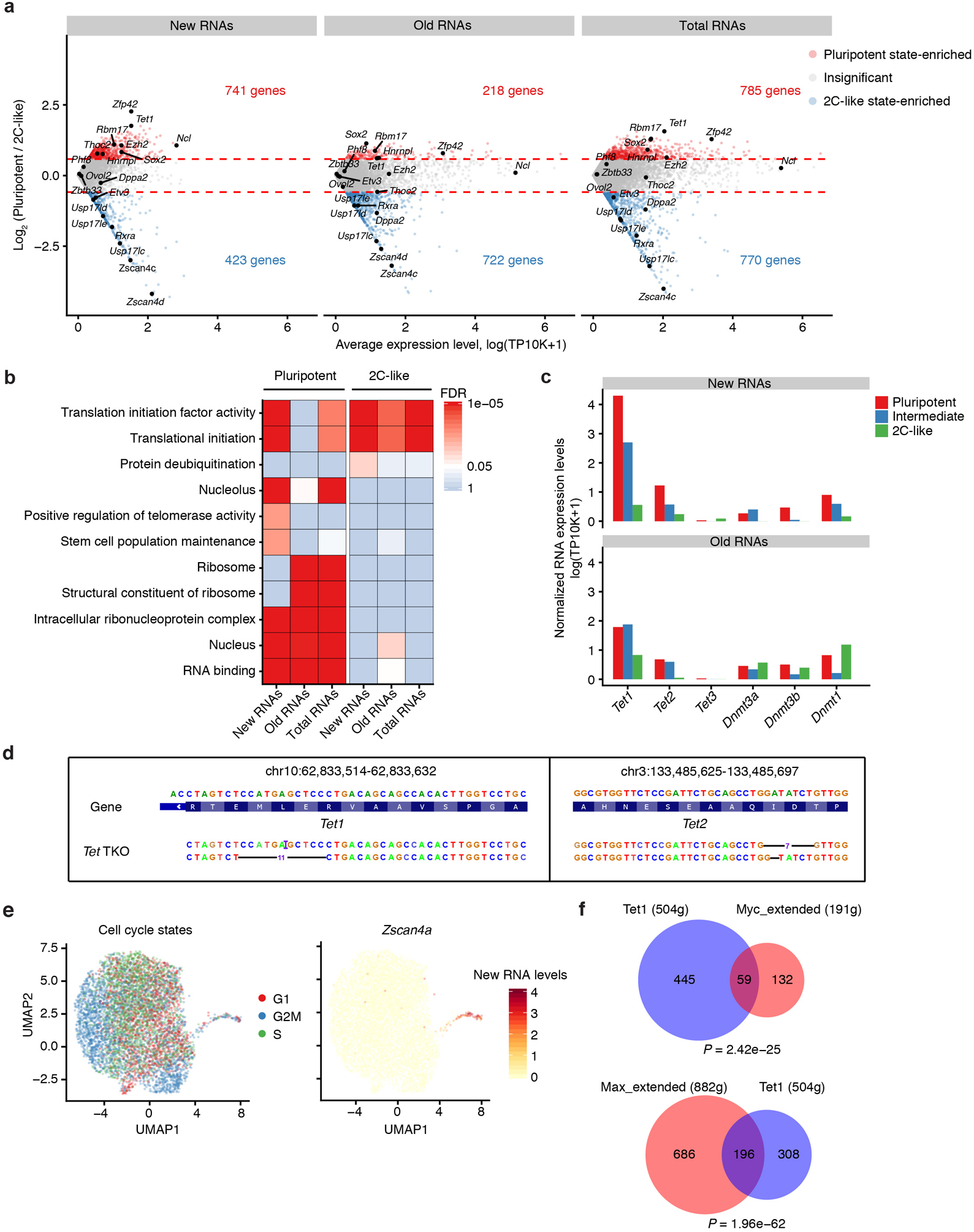
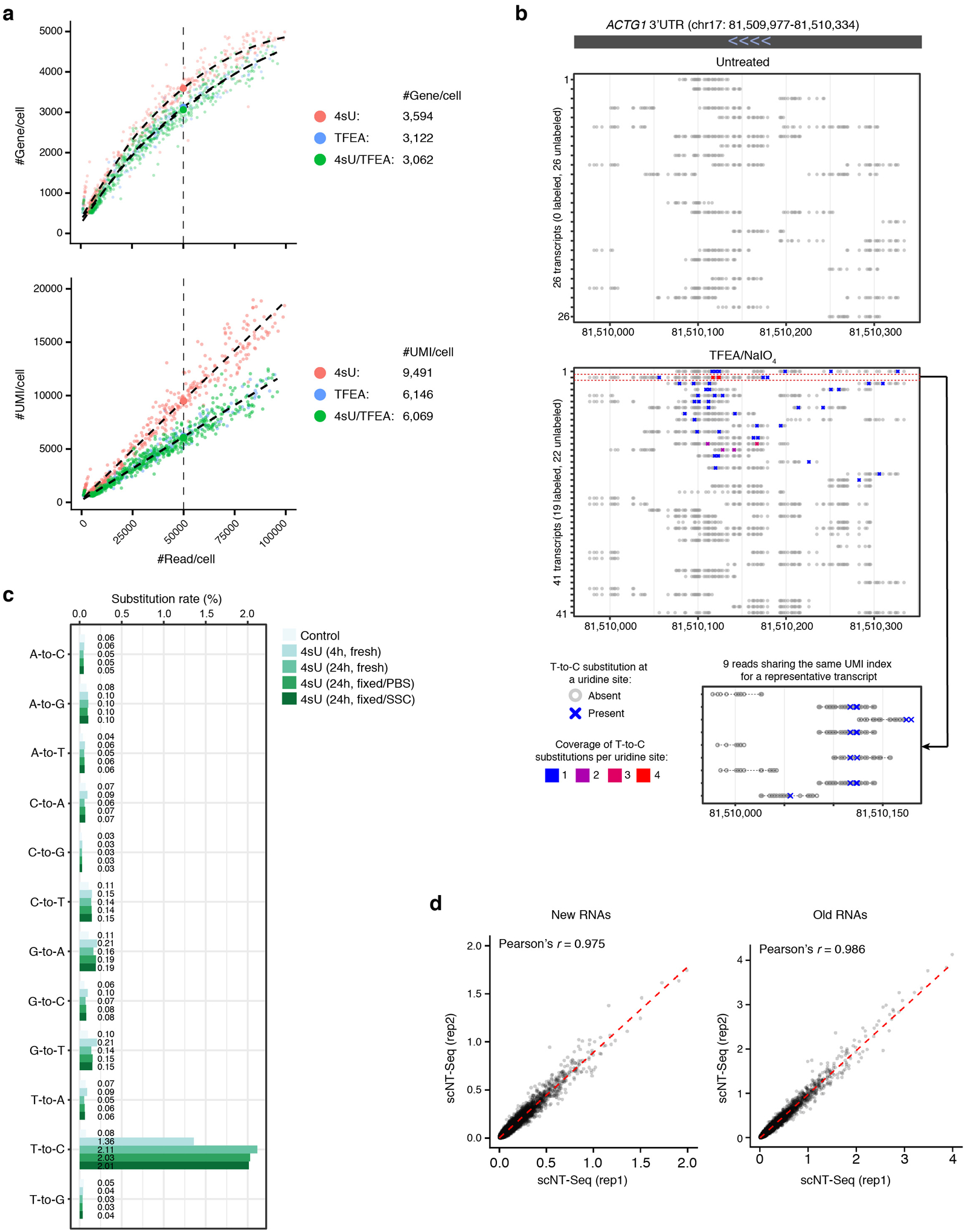
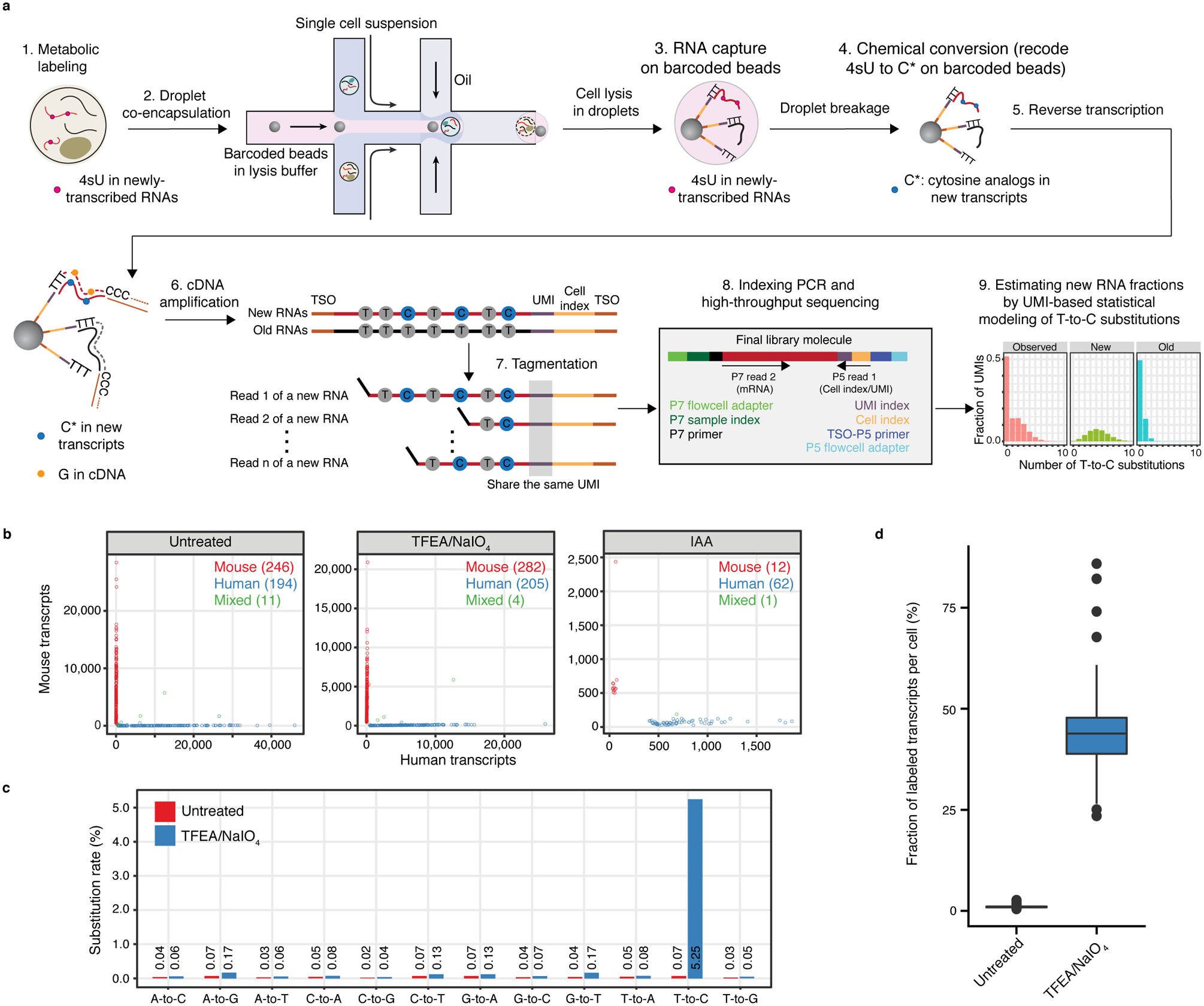
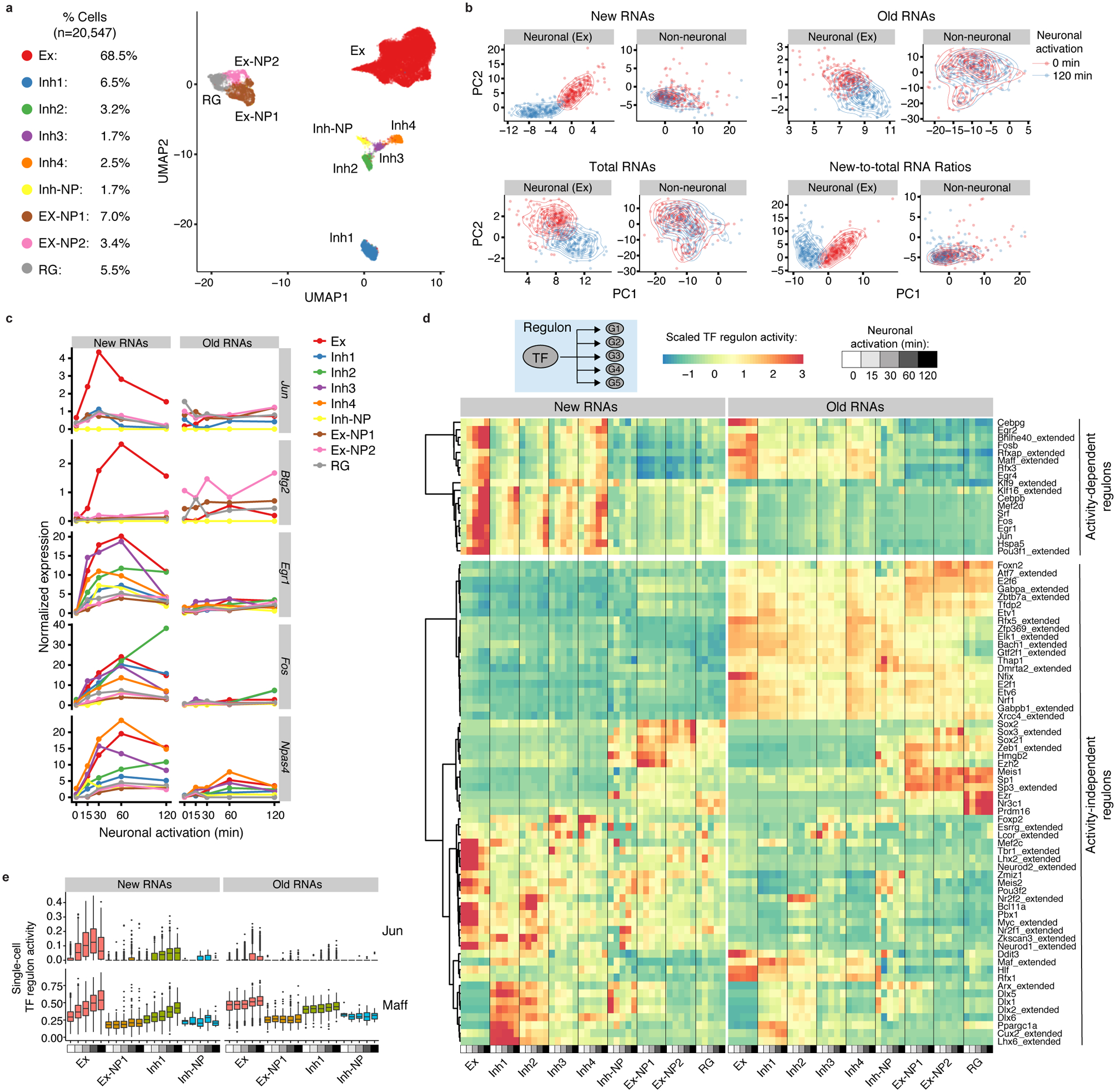
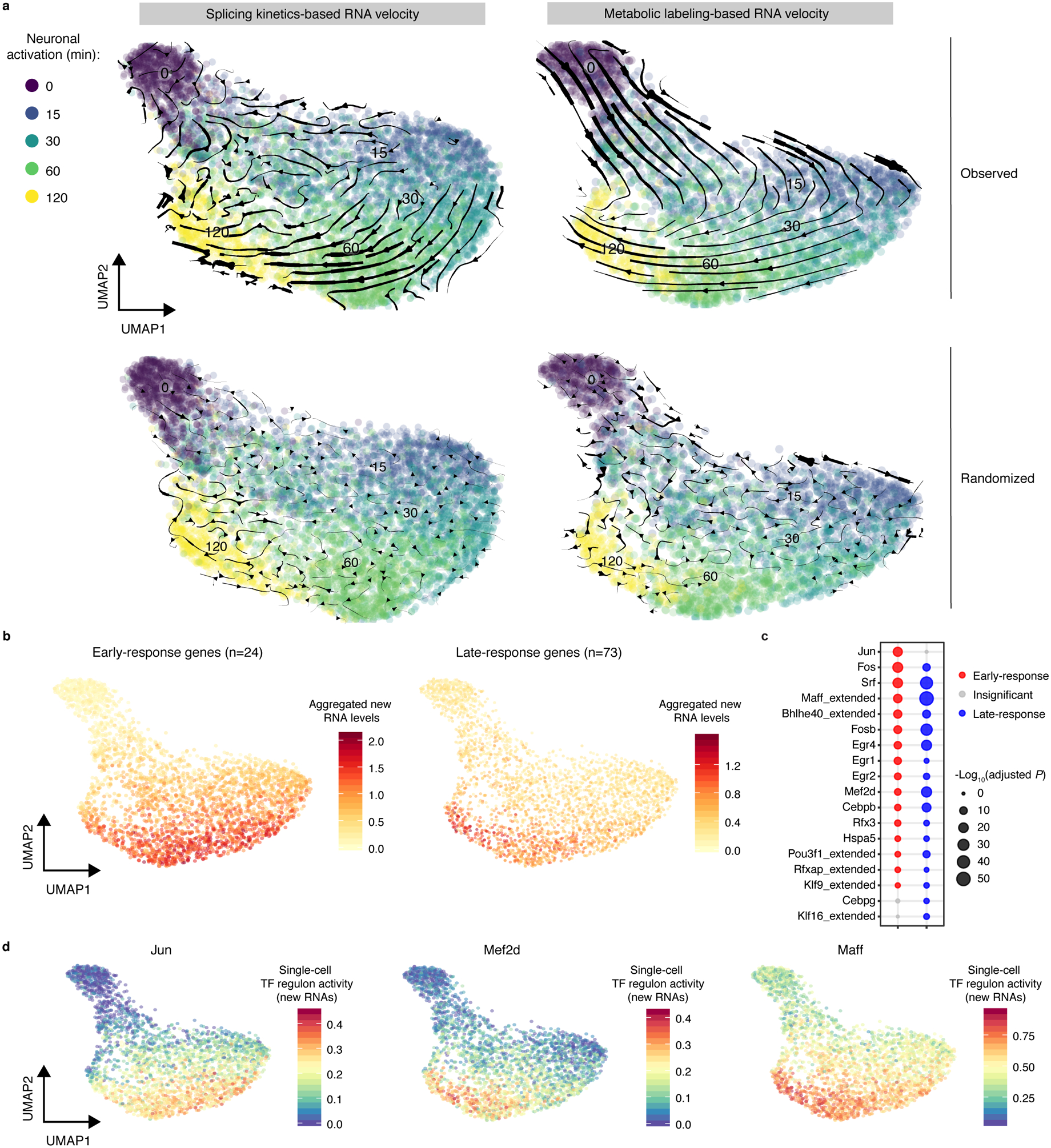
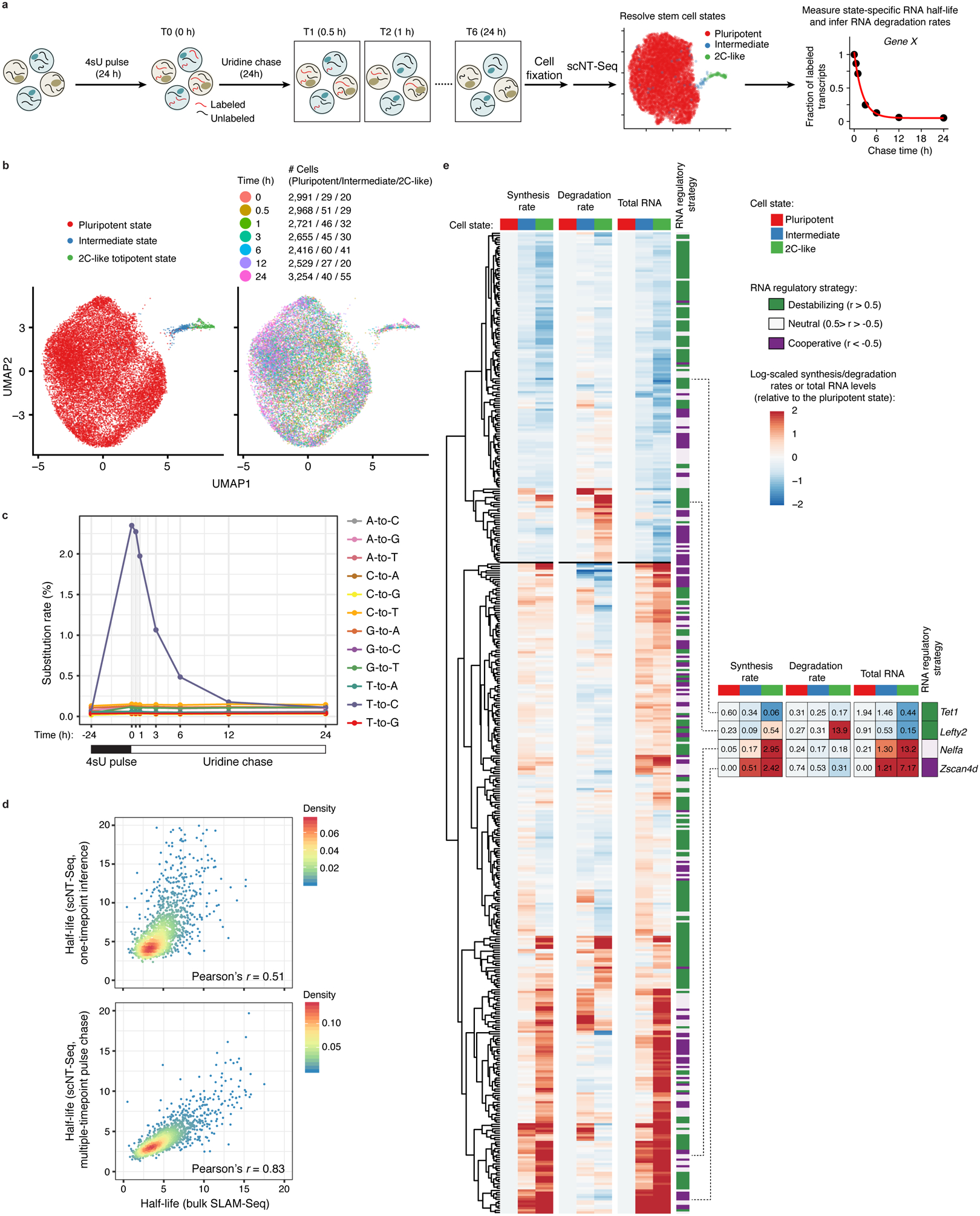
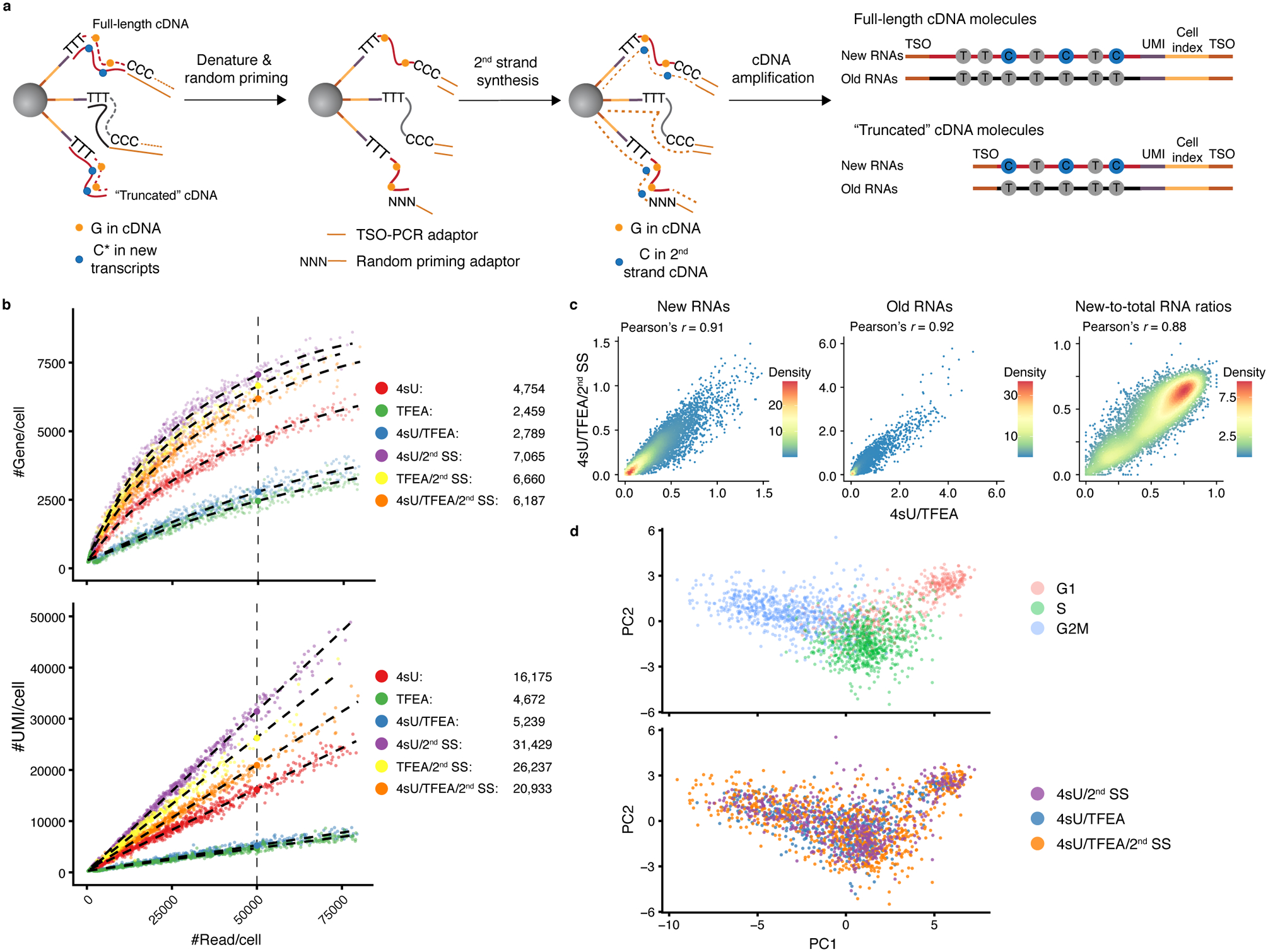
Single-cell RNA sequencing offers snapshots of whole transcriptomes but obscures the temporal RNA dynamics. Here we present single-cell metabolically labeled new RNA tagging sequencing (scNT-seq), a method for massively parallel analysis of newly transcribed and pre-existing mRNAs from the same cell. This droplet microfluidics-based method enables high-throughput chemical conversion on barcoded beads, efficiently marking newly transcribed mRNAs with T-to-C substitutions. Using scNT-seq, we jointly profiled new and old transcriptomes in ~55,000 single cells. These data revealed time-resolved transcription factor activities and cell-state trajectories at the single-cell level in response to neuronal activation. We further determined rates of RNA biogenesis and decay to uncover RNA regulatory strategies during stepwise conversion between pluripotent and rare totipotent two-cell embryo (2C)-like stem cell states. Finally, integrating scNT-seq with genetic perturbation identifies DNA methylcytosine dioxygenase as an epigenetic barrier into the 2C-like cell state. Time-resolved single-cell transcriptomic analysis thus opens new lines of inquiry regarding cell-type-specific RNA regulatory mechanisms.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures

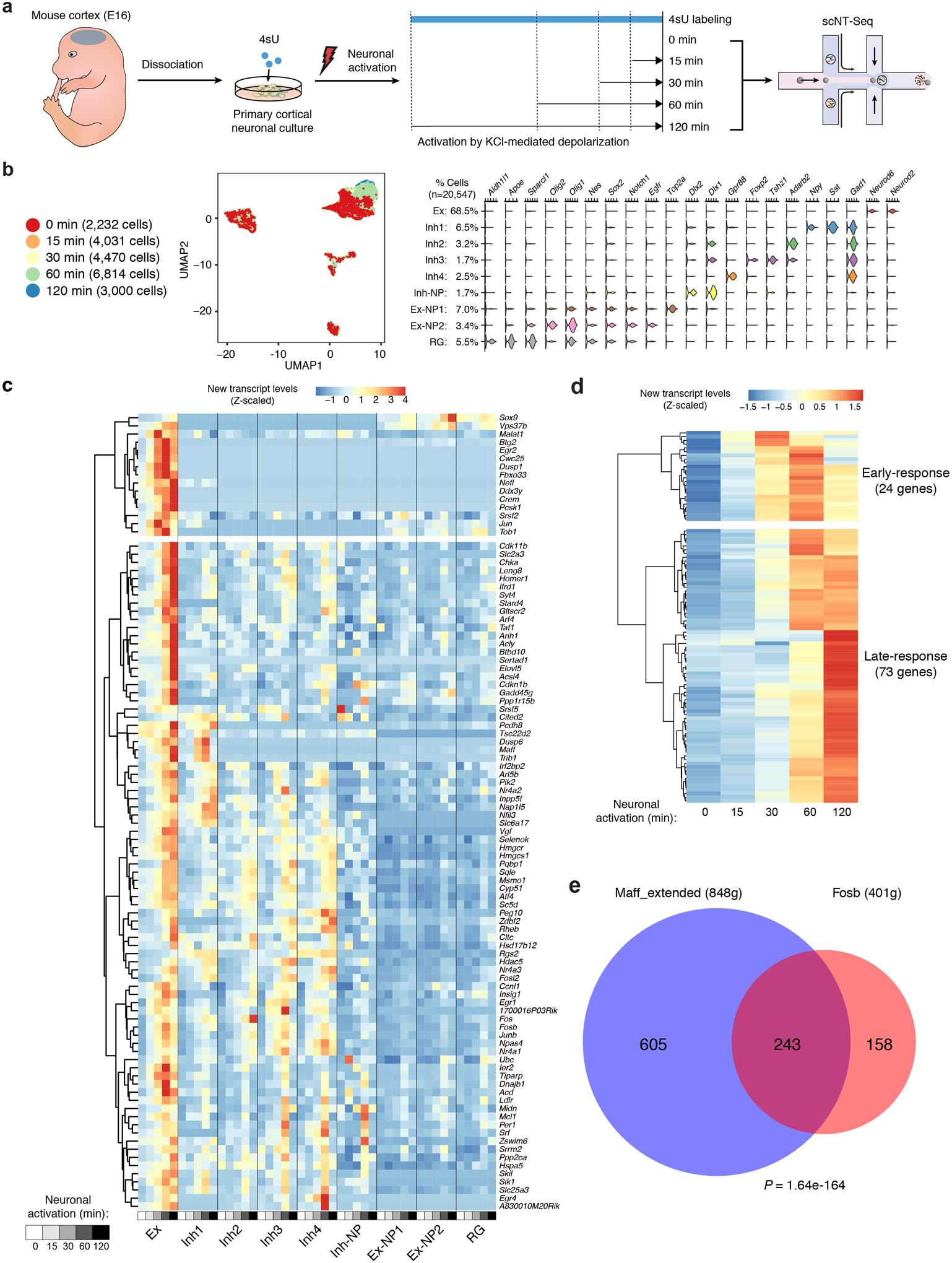
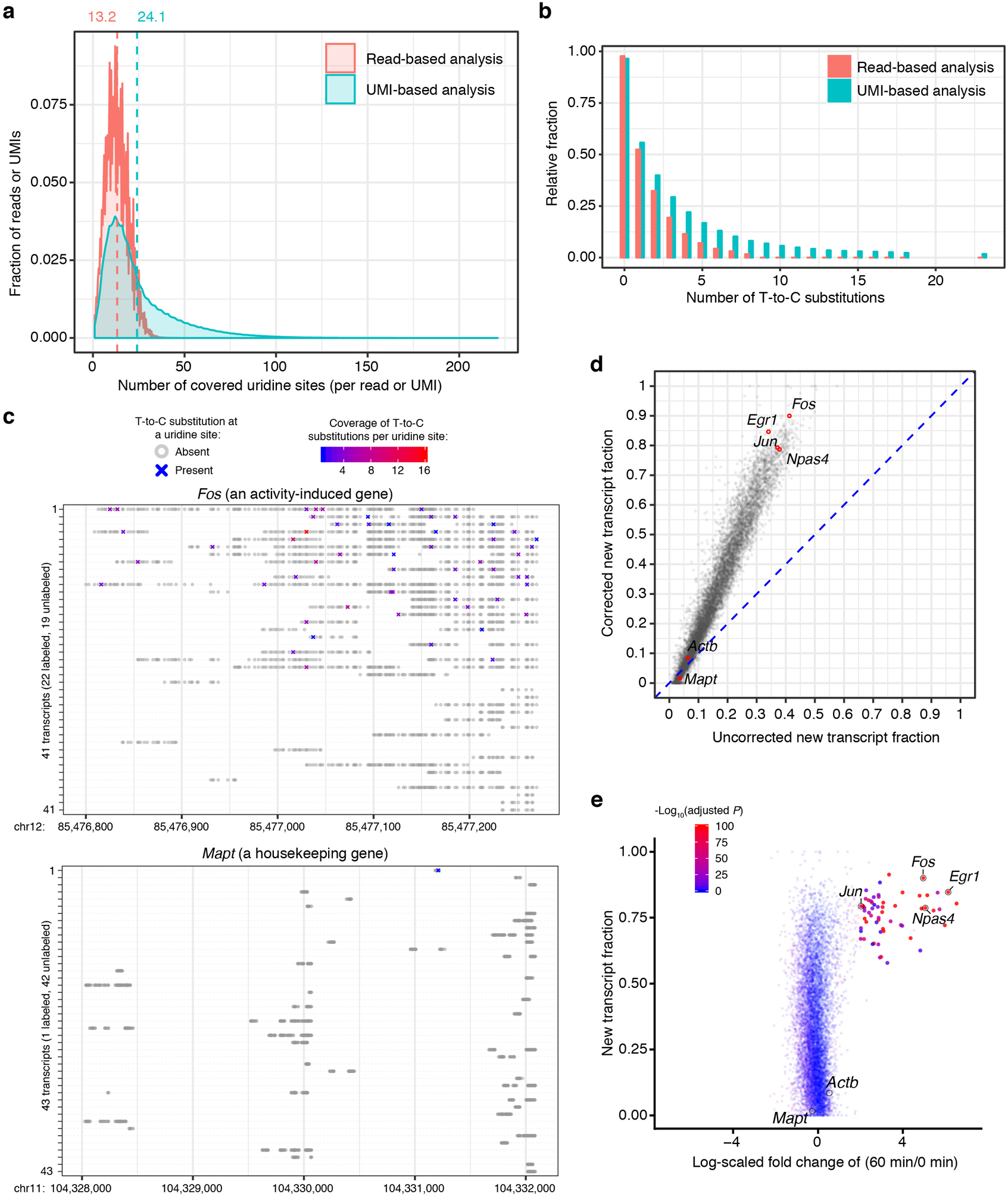

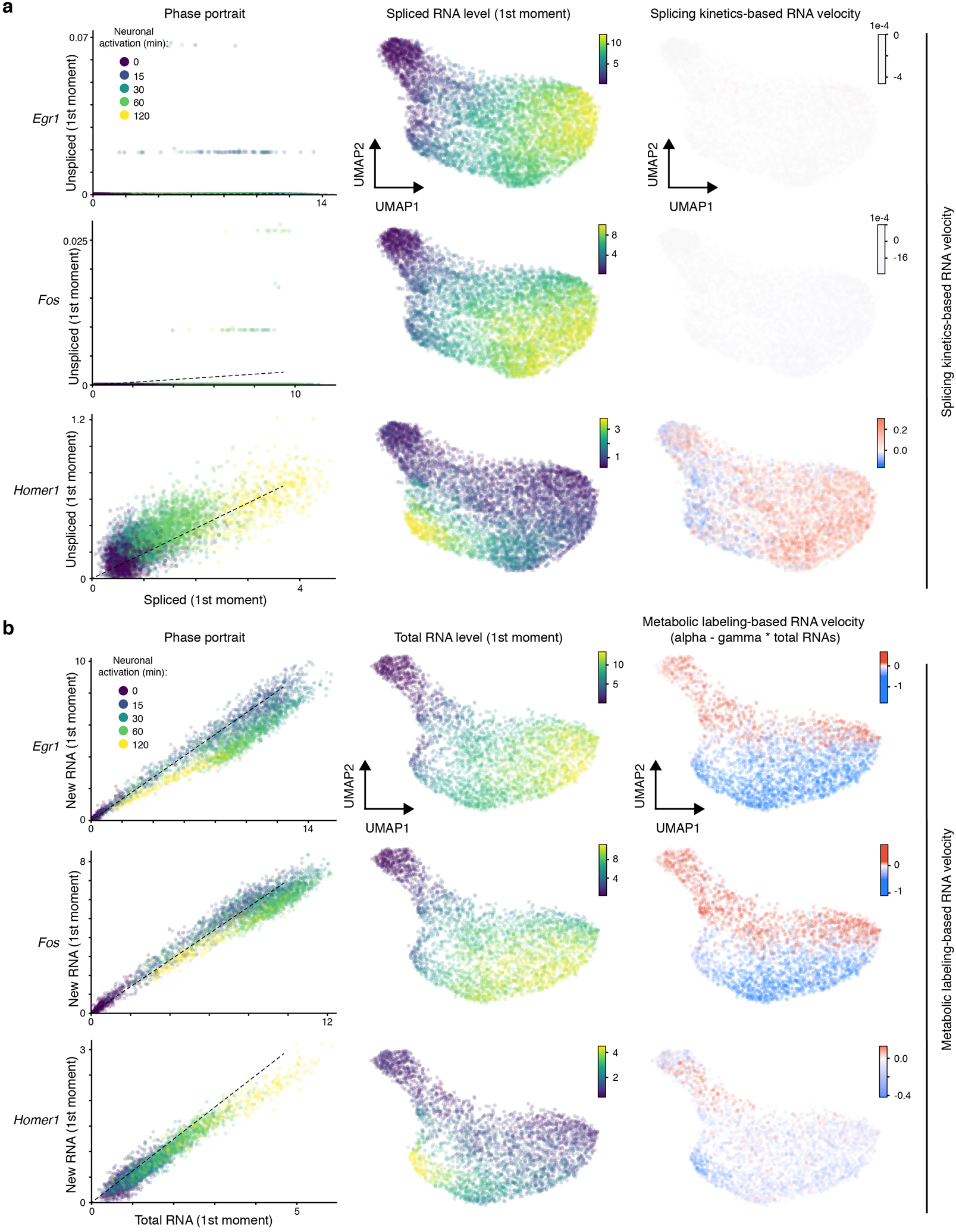
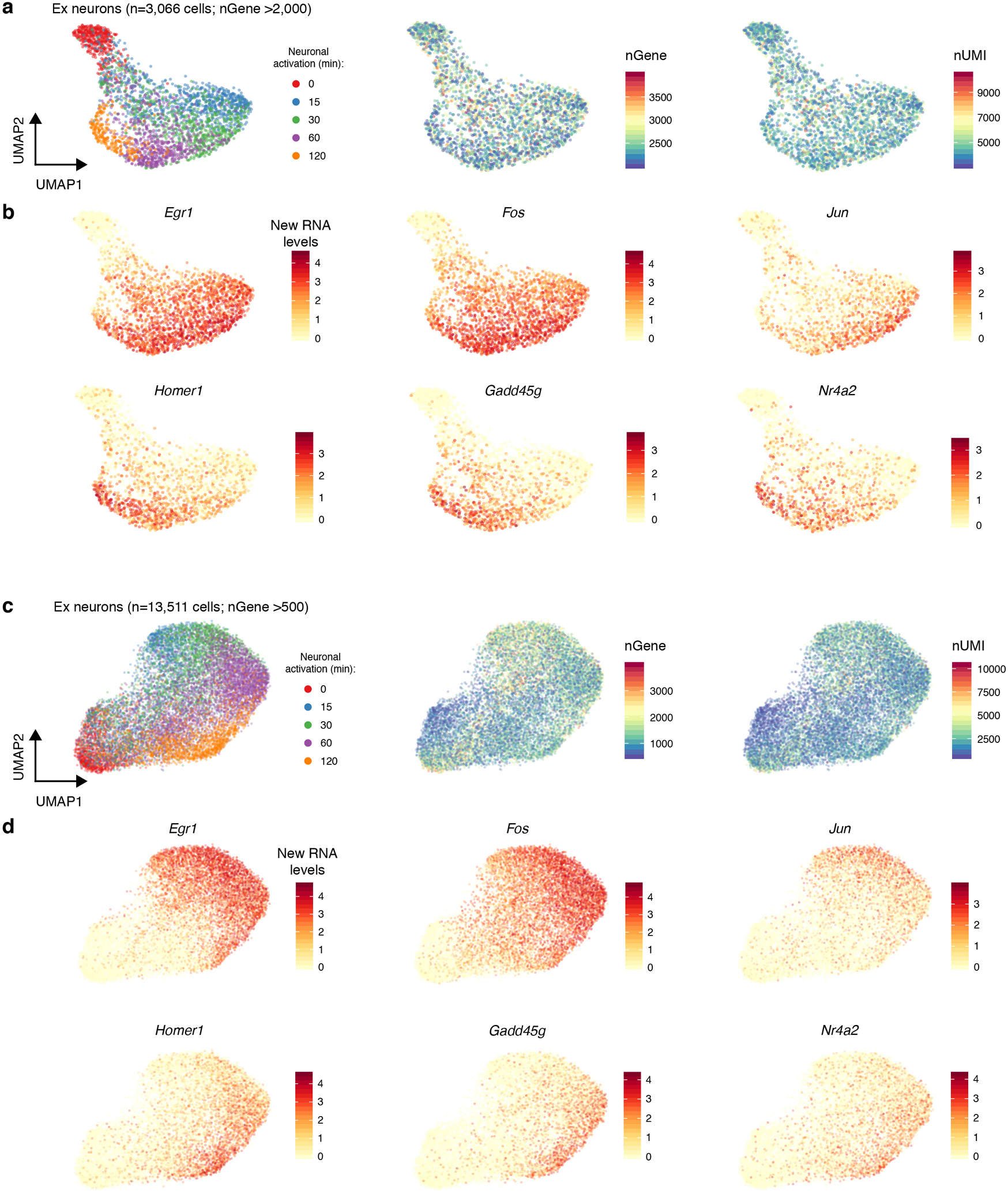

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
