

Contributions of alveolar epithelial cell quality control to pulmonary fibrosis
- PMID: 32870817
- PMCID: PMC7524463
- DOI: 10.1172/JCI139519
Contributions of alveolar epithelial cell quality control to pulmonary fibrosis
Abstract
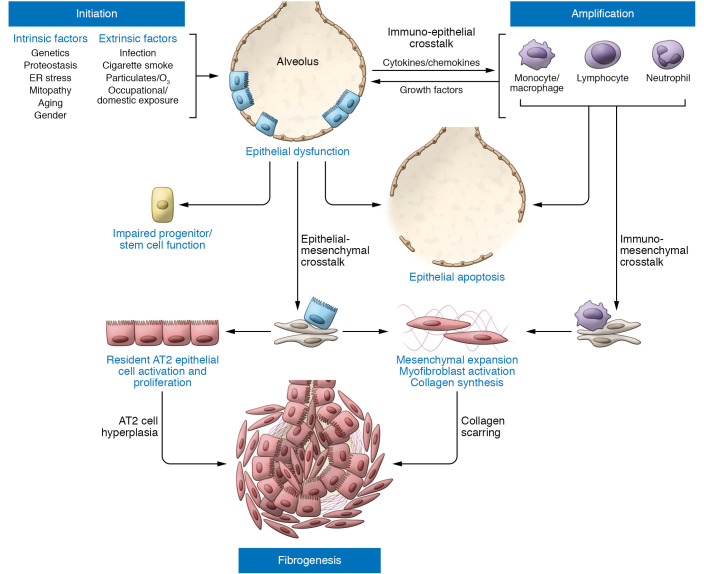
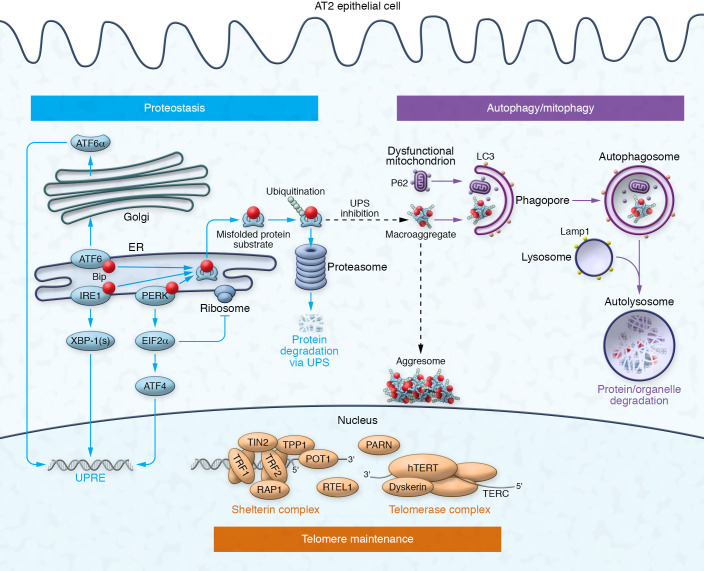
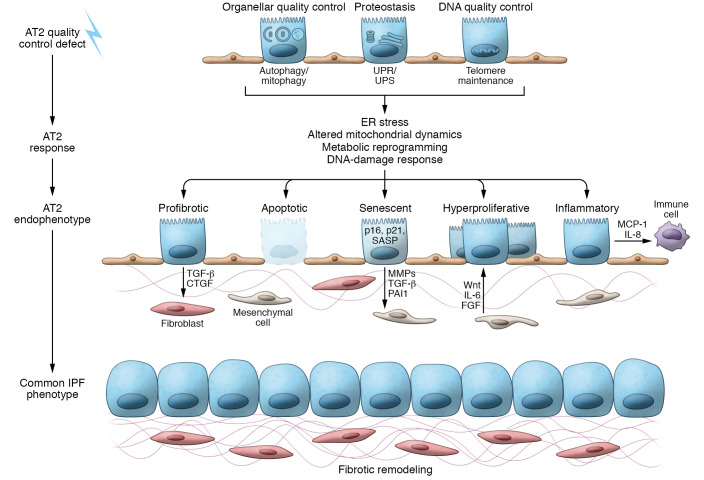
Epithelial cell dysfunction has emerged as a central component of the pathophysiology of diffuse parenchymal diseases including idiopathic pulmonary fibrosis (IPF). Alveolar type 2 (AT2) cells represent a metabolically active lung cell population important for surfactant biosynthesis and alveolar homeostasis. AT2 cells and other distal lung epithelia, like all eukaryotic cells, contain an elegant quality control network to respond to intrinsic metabolic and biosynthetic challenges imparted by mutant protein conformers, dysfunctional subcellular organelles, and dysregulated telomeres. Failed AT2 quality control components (the ubiquitin-proteasome system, unfolded protein response, macroautophagy, mitophagy, and telomere maintenance) result in diverse cellular endophenotypes and molecular signatures including ER stress, defective autophagy, mitochondrial dysfunction, apoptosis, inflammatory cell recruitment, profibrotic signaling, and altered progenitor function that ultimately converge to drive downstream fibrotic remodeling in the IPF lung. As this complex network becomes increasingly better understood, opportunities will emerge to identify targets and therapeutic strategies for IPF.
Conflict of interest statement
Figures
References
-
- Travis WD, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748. doi: 10.1164/rccm.201308-1483ST. - DOI - PMC - PubMed
-
- Chambers DC, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-sixth adult lung and heart–lung transplantation Report—2019; Focus theme: Donor and recipient size match. J Heart Lung Transplant. 2019;38(10):1042–1055. doi: 10.1016/j.healun.2019.08.001. - DOI - PMC - PubMed
