

Fibroblast growth factor homologous factors tune arrhythmogenic late NaV1.5 current in calmodulin binding-deficient channels
- PMID: 32870823
- PMCID: PMC7566708
- DOI: 10.1172/jci.insight.141736
Fibroblast growth factor homologous factors tune arrhythmogenic late NaV1.5 current in calmodulin binding-deficient channels
Abstract
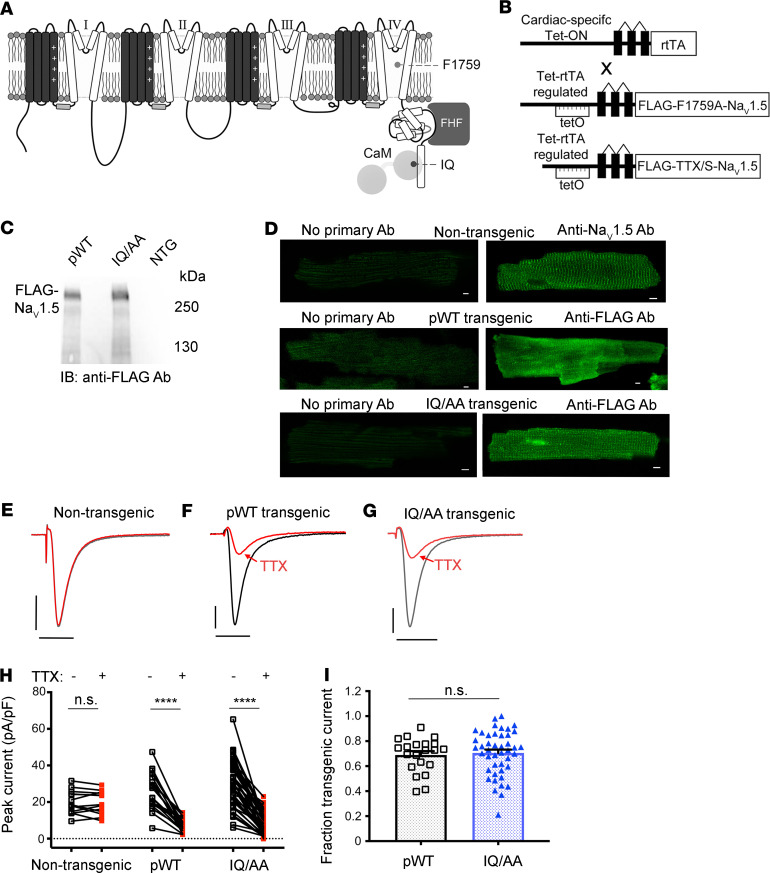
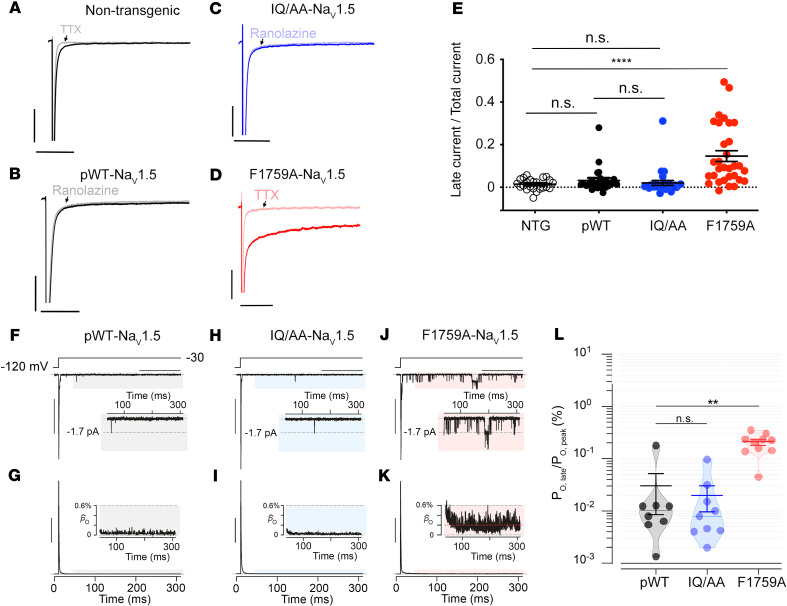
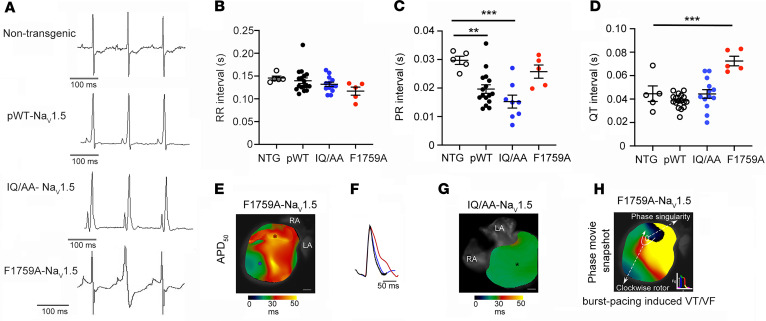
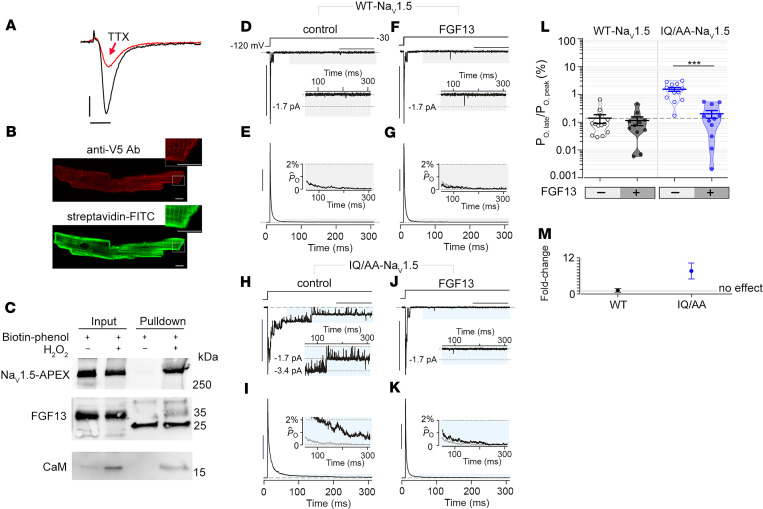
The Ca2+-binding protein calmodulin has emerged as a pivotal player in tuning Na+ channel function, although its impact in vivo remains to be resolved. Here, we identify the role of calmodulin and the NaV1.5 interactome in regulating late Na+ current in cardiomyocytes. We created transgenic mice with cardiac-specific expression of human NaV1.5 channels with alanine substitutions for the IQ motif (IQ/AA). The mutations rendered the channels incapable of binding calmodulin to the C-terminus. The IQ/AA transgenic mice exhibited normal ventricular repolarization without arrhythmias and an absence of increased late Na+ current. In comparison, transgenic mice expressing a lidocaine-resistant (F1759A) human NaV1.5 demonstrated increased late Na+ current and prolonged repolarization in cardiomyocytes, with spontaneous arrhythmias. To determine regulatory factors that prevent late Na+ current for the IQ/AA mutant channel, we considered fibroblast growth factor homologous factors (FHFs), which are within the NaV1.5 proteomic subdomain shown by proximity labeling in transgenic mice expressing NaV1.5 conjugated to ascorbate peroxidase. We found that FGF13 diminished late current of the IQ/AA but not F1759A mutant cardiomyocytes, suggesting that endogenous FHFs may serve to prevent late Na+ current in mouse cardiomyocytes. Leveraging endogenous mechanisms may furnish an alternative avenue for developing novel pharmacology that selectively blunts late Na+ current.
Keywords: Arrhythmias; Calmodulin; Cardiology; Sodium channels.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
