

Dopa-responsive dystonia caused by tyrosine hydroxylase deficiency: Three cases report and literature review
- PMID: 32872068
- PMCID: PMC7437766
- DOI: 10.1097/MD.0000000000021753
Dopa-responsive dystonia caused by tyrosine hydroxylase deficiency: Three cases report and literature review
Abstract
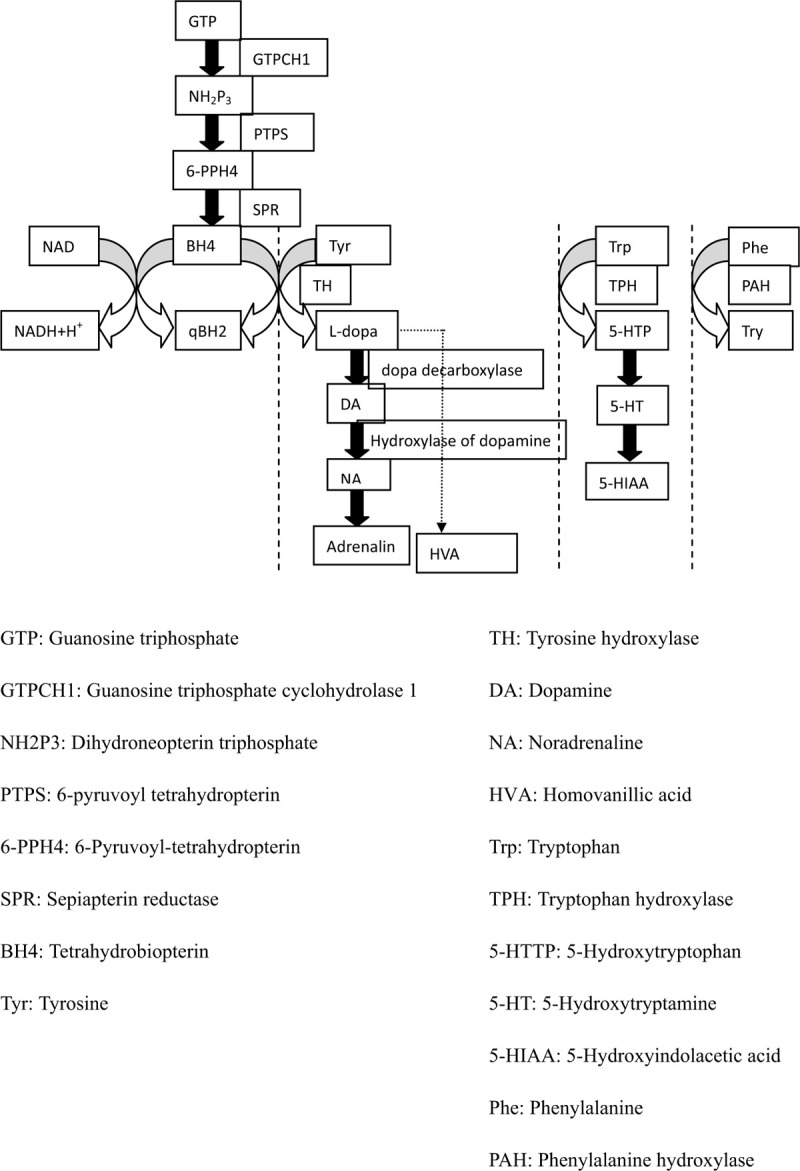
Rational: Tyrosine hydroxylase deficiency (THD) is a rare cause of dopa-responsive dystonia (DRD). Although the symptoms of DRD may be improved by treatment with L-dopa, the low morbidity of THD can lead to its misdiagnosis. Thus, it is important for physicians to be aware of THD as a cause of DRD.
Patient concerns: We report 3 cases of THD. A 5-year-old boy with DRD was diagnosed with THD and found to have compound heterozygous mutations of the TH gene, including TH:c.647G>C from his mother and TH:c.646G>A from his father. Two female siblings also were found to have TH:c.698G>A from their mother and TH:c.710T>C from their father. The younger daughter, at age 3.5 years, was diagnosed with DRD caused by THD, and then the diagnosis of the older daughter, at age 11 years, was changed from cerebral palsy to DRD caused by THD.
Diagnosis: The diagnosis of dopa-responsive dystonia caused by tyrosine hydroxylase deficiency was determined by whole exome sequencing.
Intervention: They all treated with low dose levodopa and benserazide tablets.
Outcomes: The boy had a very good therapeutic effect, and he could walk very well by the second day of treatment. The younger sister of the siblings had a partial therapeutic effect, but her elder sister was only little effective with a milder improvement of dystonia and improvement of myodynamia.
Conclusion: The characteristics of THD are heterogeneous, and its phenotypes are classified as type A or type B according to increasing severity. Generally, L-dopa has a good therapeutic effect in cases with type A phenotypes. We reviewed 87 cases of reported in the literature and found that c.698G>A and c.707T>C are hot spot mutations. Changes on cerebral magnetic resonance imaging were nonspecific. Analysis of neurotransmitter levels in cerebrospinal fluid is an invasive means of achieving a biochemical diagnosis.
Conflict of interest statement
The authors have no conflicts of interests to disclose.
Figures
References
-
- Segawa M, Hoosaks A, Miyagawa F, et al. Hereditary progressive dystonia with marked diurnal fluctuation. Adv Neurol 1976;14:215–33. - PubMed
-
- Nygaard TG, Marsden CD, Duvoisin RC. Dopa responsive dystonia. Adv Neurol 1988;50:377–84. - PubMed
-
- Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol 2003;54:32–45. - PubMed
-
- Ichinose H, Ohye T, Takahashi E, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994;8:236–42. - PubMed
-
- Knappskog PM, Flatmark T, Mallet J, et al. Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum Mol Genet 1995;4:1209. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
