

Neutralization of Lipocalin-2 Diminishes Stroke-Reperfusion Injury
- PMID: 32872405
- PMCID: PMC7503651
- DOI: 10.3390/ijms21176253
Neutralization of Lipocalin-2 Diminishes Stroke-Reperfusion Injury
Abstract
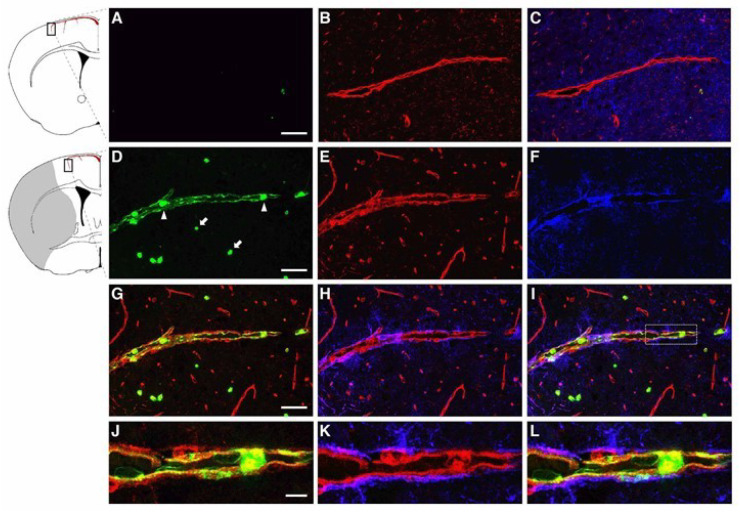
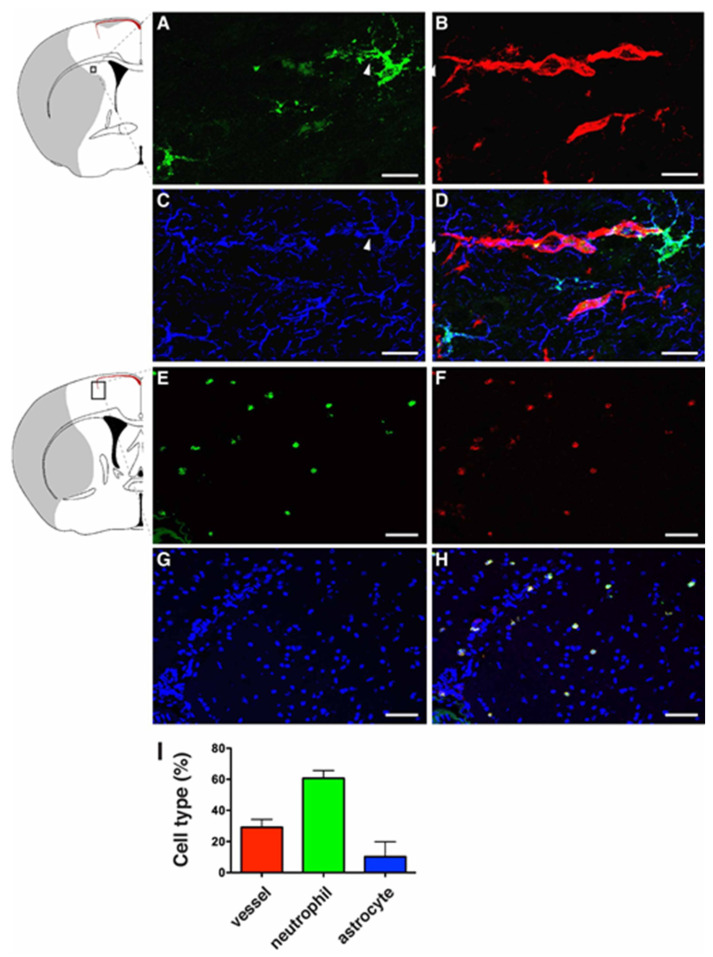
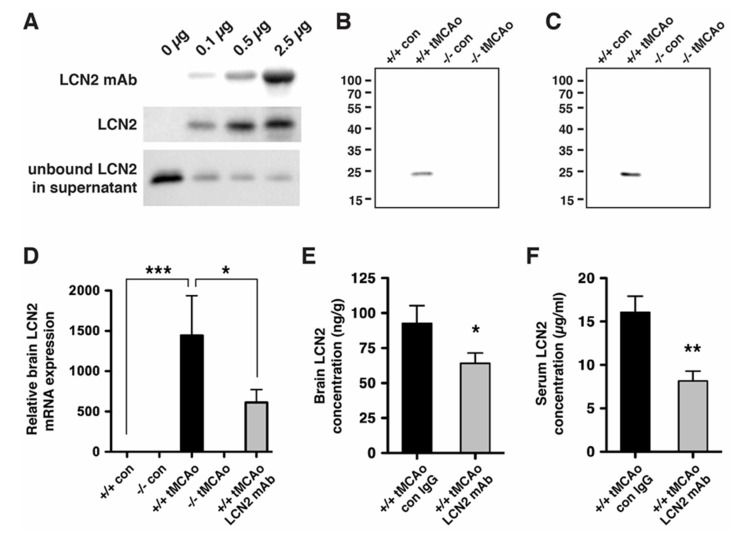
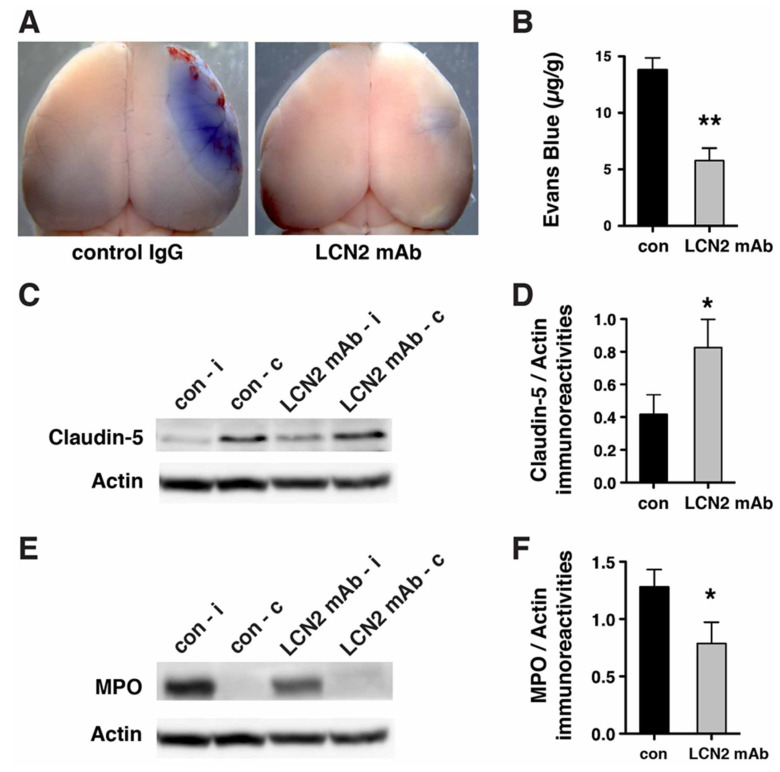
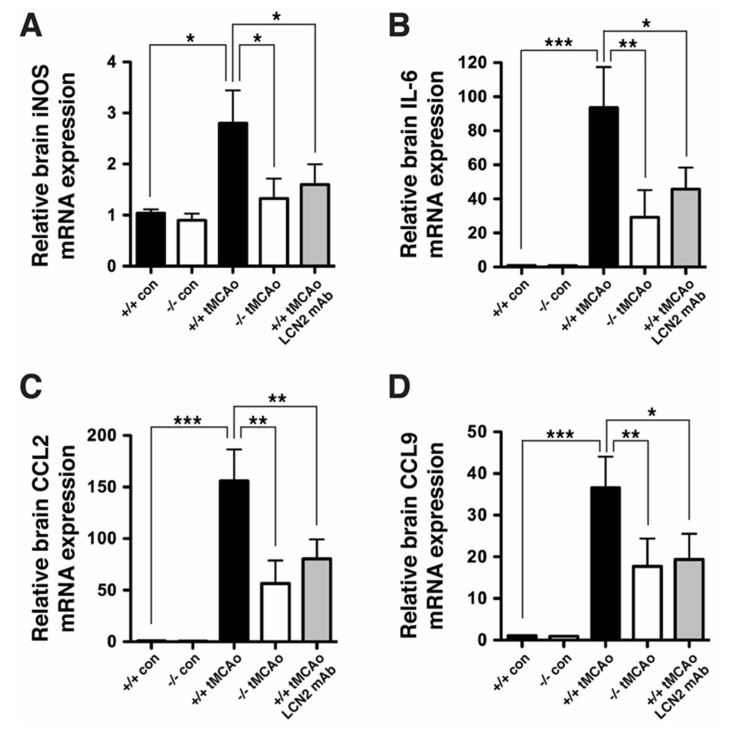
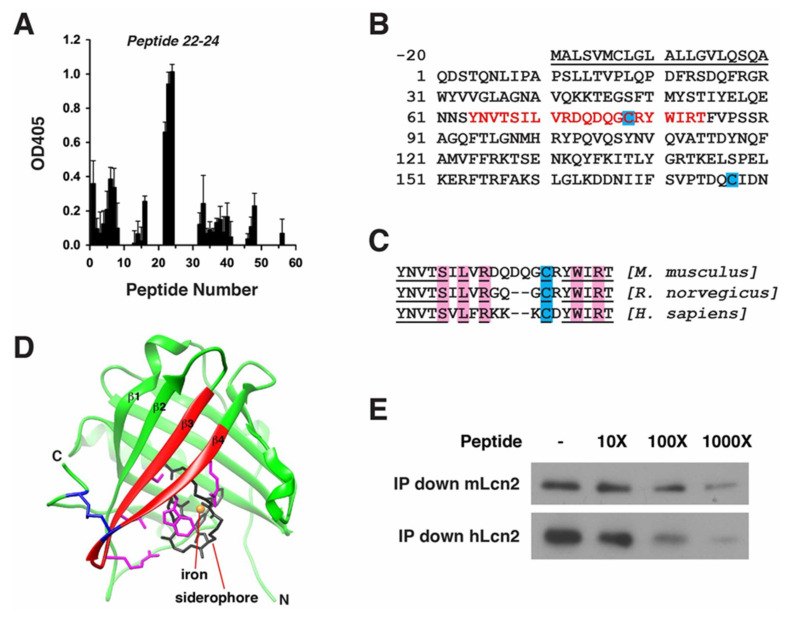
Oxidative stress is a key contributor to the pathogenesis of stroke-reperfusion injury. Neuroinflammatory peptides released after ischemic stroke mediate reperfusion injury. Previous studies, including ours, have shown that lipocalin-2 (LCN2) is secreted in response to cerebral ischemia to promote reperfusion injury. Genetic deletion of LCN2 significantly reduces brain injury after stroke, suggesting that LCN2 is a mediator of reperfusion injury and a potential therapeutic target. Immunotherapy has the potential to harness neuroinflammatory responses and provides neuroprotection against stroke. Here we report that LCN2 was induced on the inner surface of cerebral endothelial cells, neutrophils, and astrocytes that gatekeep the blood-brain barrier (BBB) after stroke. LCN2 monoclonal antibody (mAb) specifically targeted LCN2 in vitro and in vivo, attenuating the induction of LCN2 and pro-inflammatory mediators (iNOS, IL-6, CCL2, and CCL9) after stroke. Administration of LCN2 mAb at 4 h after stroke significantly reduced neurological deficits, cerebral infarction, edema, BBB leakage, and infiltration of neutrophils. The binding epitope of LCN2 mAb was mapped to the β3 and β4 strands, which are responsible for maintaining the integrity of LCN2 cup-shaped structure. These data indicate that LCN2 can be pharmacologically targeted using a specific mAb to reduce reperfusion injury after stroke.
Keywords: Lipocalin-2; immunotherapy; neutrophils; oxidative stress; reperfusion injury; stroke.
Conflict of interest statement
The authors declare that no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures

References
-
- Krishnamurthi R.V., Feigin V.L., Forouzanfar M.H., Mensah G.A., Connor M., Bennett D.A., Moran A.E., Sacco R.L., Anderson L.M., Truelsen T., et al. Global and regional burden of first-ever ischaemic and haemorrhagic stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet Glob. Health. 2013;1:e259–e281. doi: 10.1016/S2214-109X(13)70089-5. - DOI - PMC - PubMed
-
- Campbell B.C.V., Donnan G.A., Lees K.R., Hacke W., Khatri P., Hill M.D., Goyal M., Mitchell P.J., Saver J.L., Diener H.C., et al. Endovascular stent thrombectomy: The new standard of care for large vessel ischaemic stroke. Lancet Neurol. 2015;14:846–854. doi: 10.1016/S1474-4422(15)00140-4. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
