

Pathophysiology of Type 2 Diabetes Mellitus
- PMID: 32872570
- PMCID: PMC7503727
- DOI: 10.3390/ijms21176275
Pathophysiology of Type 2 Diabetes Mellitus
Abstract
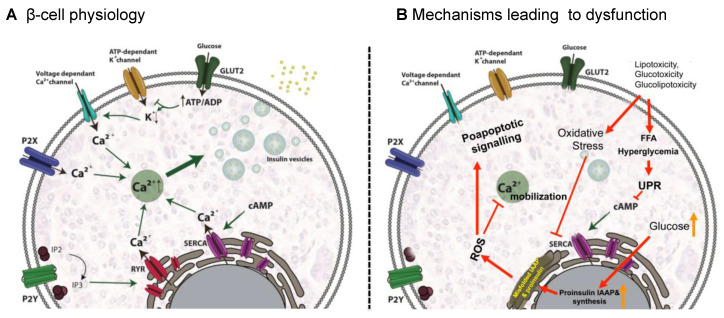
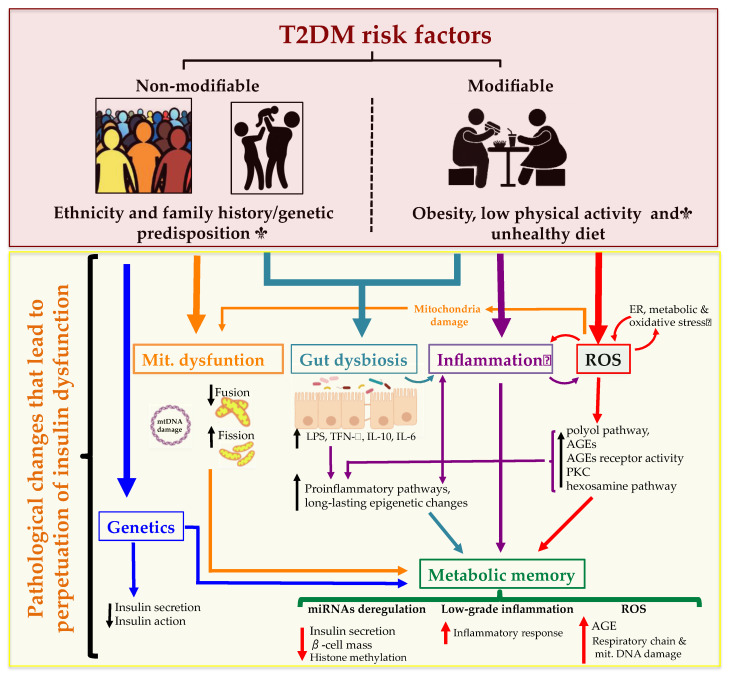
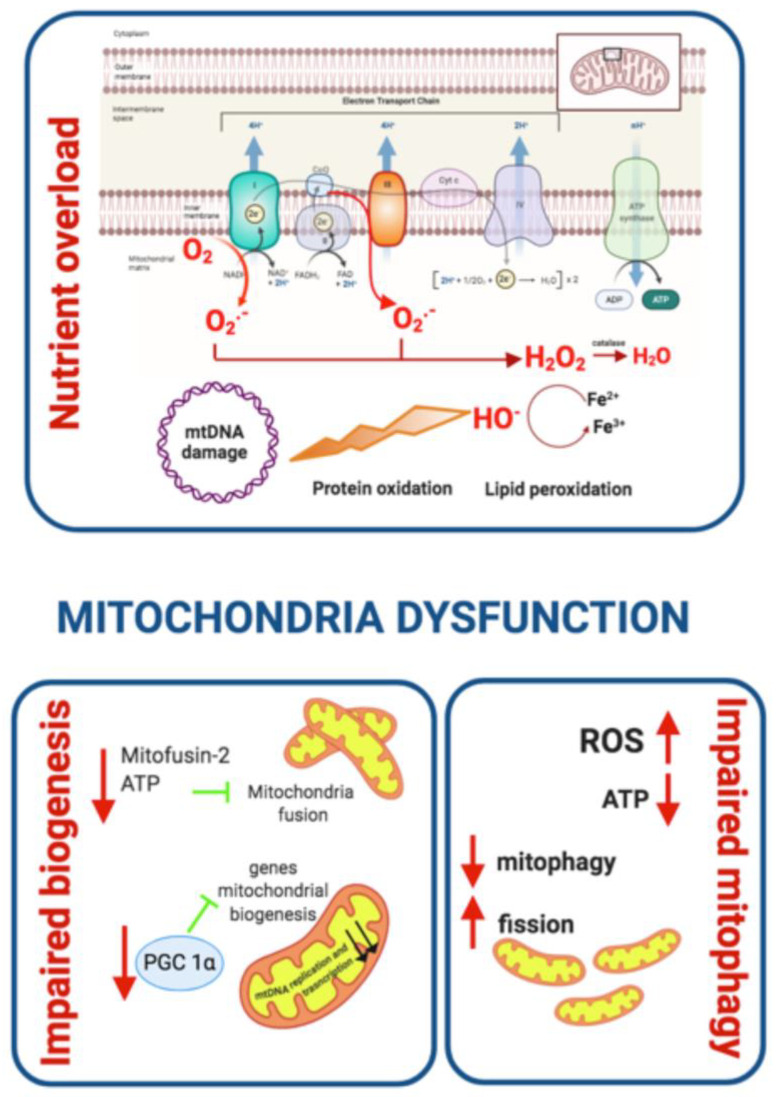
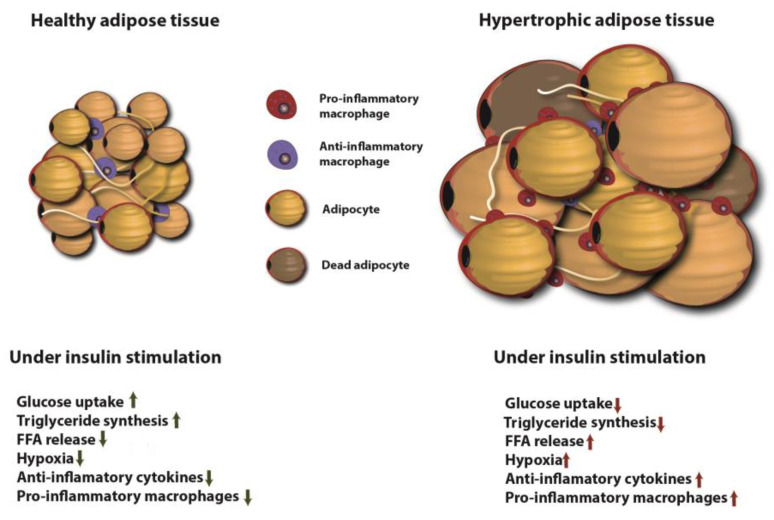
Type 2 Diabetes Mellitus (T2DM), one of the most common metabolic disorders, is caused by a combination of two primary factors: defective insulin secretion by pancreatic β-cells and the inability of insulin-sensitive tissues to respond appropriately to insulin. Because insulin release and activity are essential processes for glucose homeostasis, the molecular mechanisms involved in the synthesis and release of insulin, as well as in its detection are tightly regulated. Defects in any of the mechanisms involved in these processes can lead to a metabolic imbalance responsible for the development of the disease. This review analyzes the key aspects of T2DM, as well as the molecular mechanisms and pathways implicated in insulin metabolism leading to T2DM and insulin resistance. For that purpose, we summarize the data gathered up until now, focusing especially on insulin synthesis, insulin release, insulin sensing and on the downstream effects on individual insulin-sensitive organs. The review also covers the pathological conditions perpetuating T2DM such as nutritional factors, physical activity, gut dysbiosis and metabolic memory. Additionally, because T2DM is associated with accelerated atherosclerosis development, we review here some of the molecular mechanisms that link T2DM and insulin resistance (IR) as well as cardiovascular risk as one of the most important complications in T2DM.
Keywords: adipocyte; cardiovascular disease; insulin resistance; liver; muscle; pathophysiology; type 2 diabetes mellitus; β-cell.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
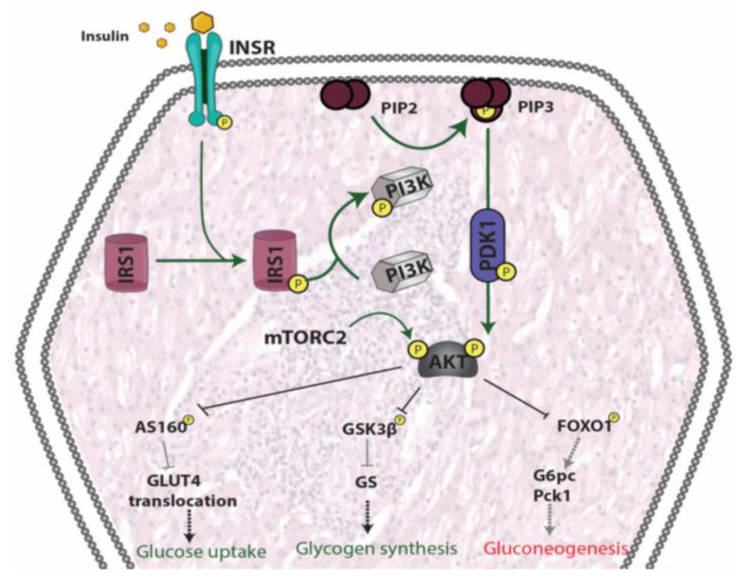
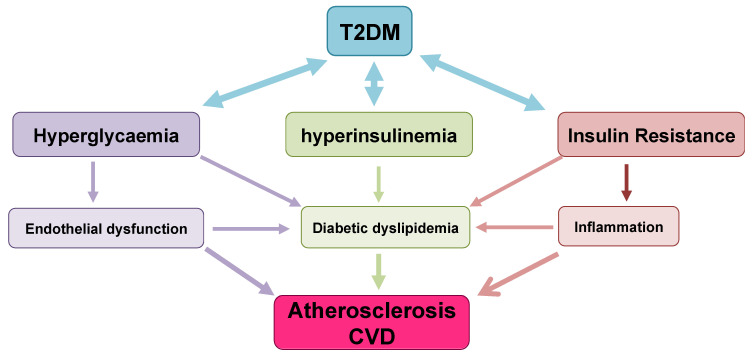
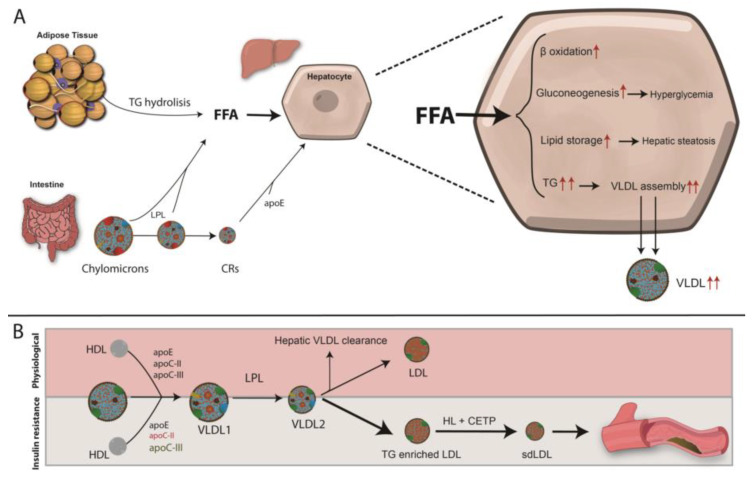
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
