

Inhibitors of BRAF dimers using an allosteric site
- PMID: 32873792
- PMCID: PMC7462985
- DOI: 10.1038/s41467-020-18123-2
Inhibitors of BRAF dimers using an allosteric site
Abstract
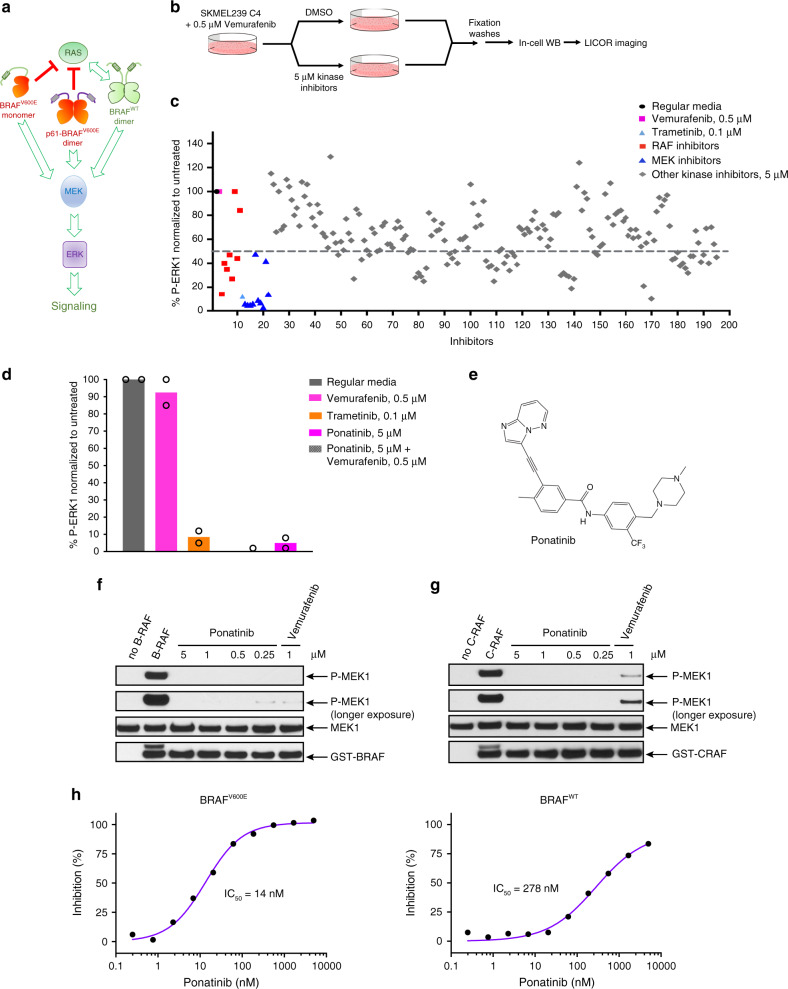
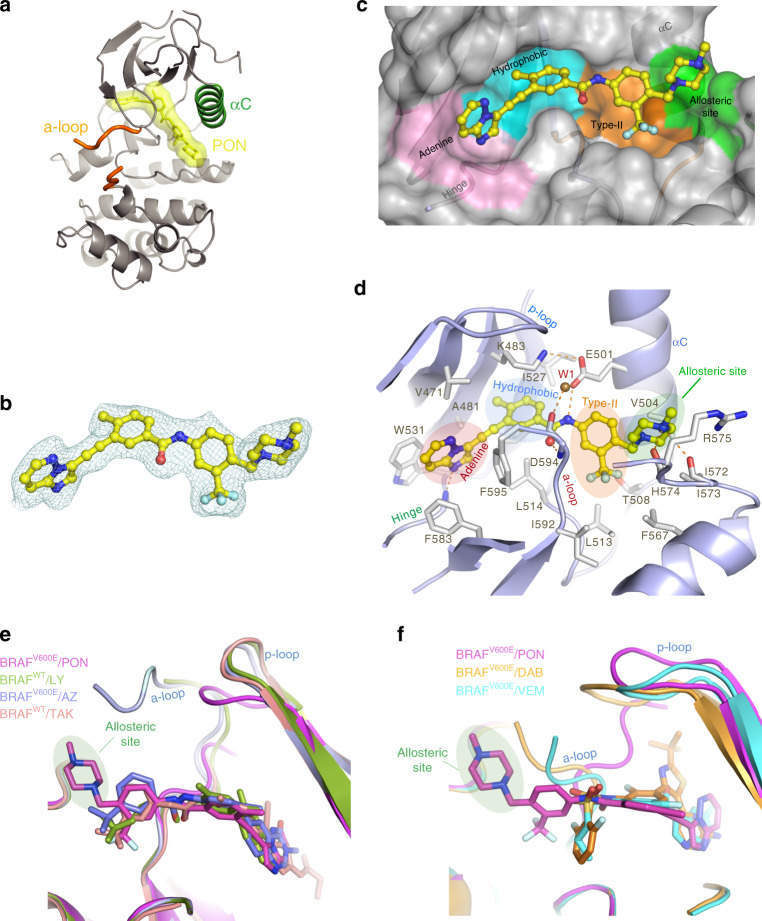
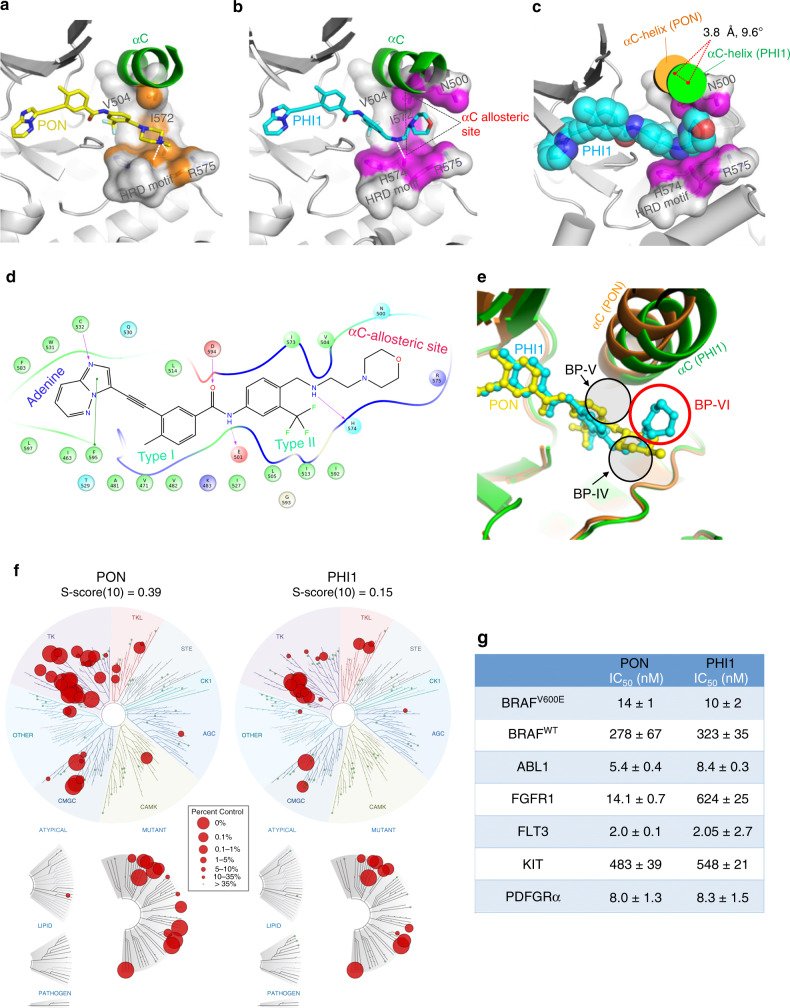
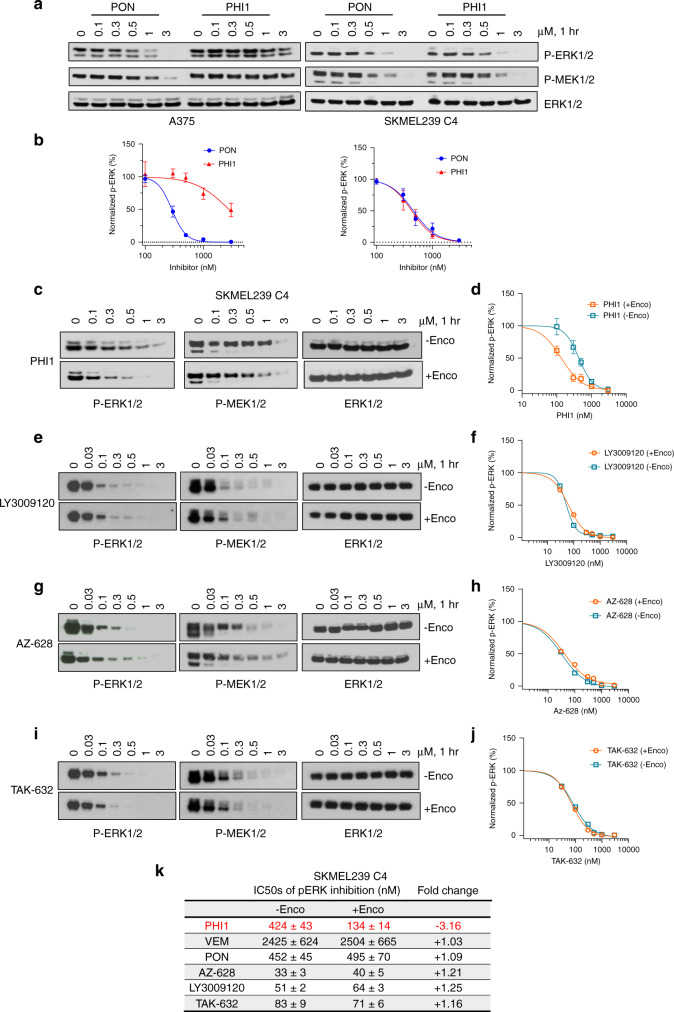
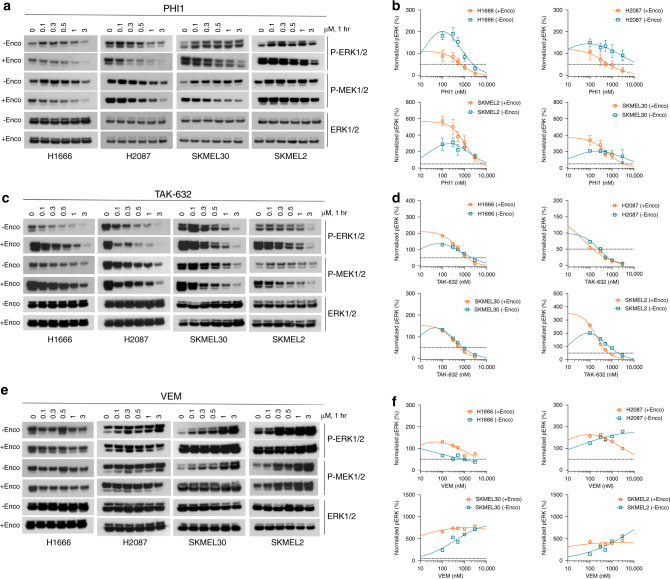
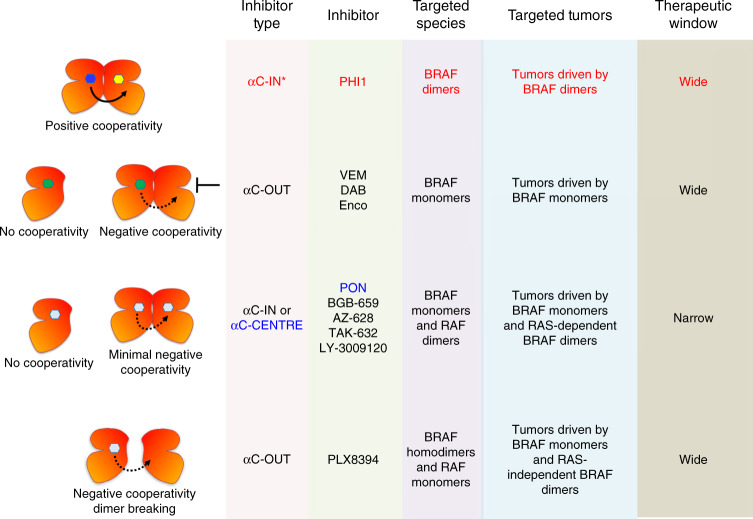
BRAF kinase, a critical effector of the ERK signaling pathway, is hyperactivated in many cancers. Oncogenic BRAFV600E signals as an active monomer in the absence of active RAS, however, in many tumors BRAF dimers mediate ERK signaling. FDA-approved RAF inhibitors poorly inhibit BRAF dimers, which leads to tumor resistance. We found that Ponatinib, an FDA-approved drug, is an effective inhibitor of BRAF monomers and dimers. Ponatinib binds the BRAF dimer and stabilizes a distinct αC-helix conformation through interaction with a previously unrevealed allosteric site. Using these structural insights, we developed PHI1, a BRAF inhibitor that fully uncovers the allosteric site. PHI1 exhibits discrete cellular selectivity for BRAF dimers, with enhanced inhibition of the second protomer when the first protomer is occupied, comprising a novel class of dimer selective inhibitors. This work shows that Ponatinib and BRAF dimer selective inhibitors will be useful in treating BRAF-dependent tumors.
Conflict of interest statement
E.G. and B.A. are inventors to US patent applications no. 62/810,056 and no. 62/810,799 filled by Albert Einstein College of Medicine that cover compounds and compositions to treat cancer related to this manuscript. E.G. has ownership interest in Stelexis Therapeutics, Selphagy Therapeutics, Aspida Therapeutics, and is a consultant/advisory board member for Stelexis Therapeutics, Selphagy Therapeutics, and Life Biosciences. None of the above companies have financial interest in this study or sponsored this research. No competing interests were disclosed by the other authors.
Figures


References
-
- Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015;16:281–298. - PubMed
-
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005;6:827–837. - PubMed
-
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004;5:875–885. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
