

Two Cases of Recessive Intellectual Disability Caused by NDST1 and METTL23 Variants
- PMID: 32878022
- PMCID: PMC7563614
- DOI: 10.3390/genes11091021
Two Cases of Recessive Intellectual Disability Caused by NDST1 and METTL23 Variants
Abstract
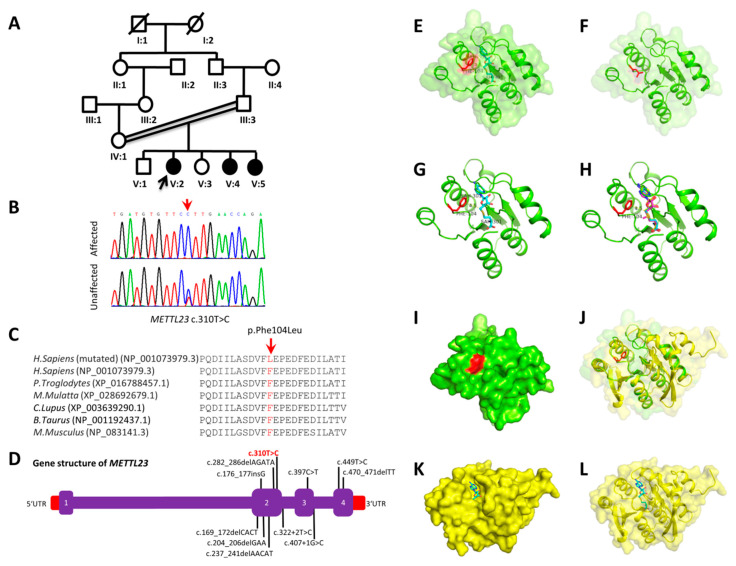
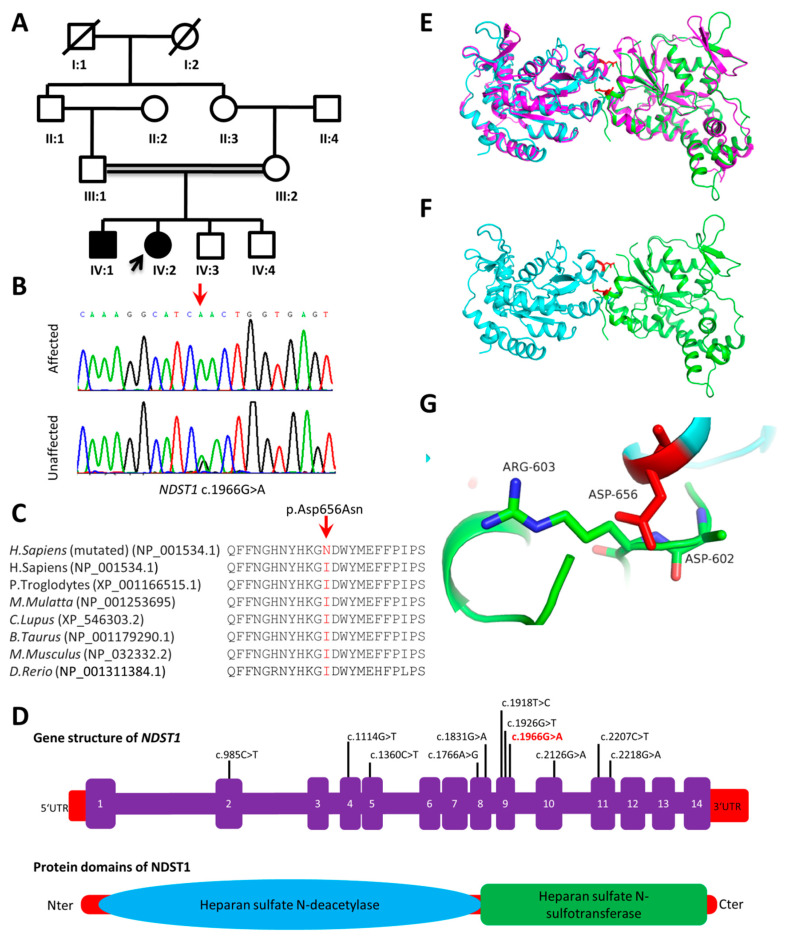
Intellectual disability (ID) is a highly heterogeneous genetic condition with more than a thousand genes described so far. By exome sequencing of two consanguineous families presenting hallmark features of ID, we identified two homozygous variants in two genes previously associated with autosomal recessive ID: NDST1 (c.1966G>A; p.Asp656Asn) and METTL23 (c.310T>C; p.Phe104Leu). The segregation of the variants was validated by Sanger sequencing in all family members. In silico homology modeling of wild-type and mutated proteins revealed substantial changes in the secondary structure of both proteins, indicating a possible effect on function. The identification and validation of new pathogenic NDST1 and METTL23 variants in two cases of autosomal recessive ID further highlight the importance of these genes in proper brain function and development.
Keywords: METTL23; NDST1; autosomal recessive intellectual disability; exome sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Rauch A., Hoyer J., Guth S., Zweier C., Kraus C., Becker C., Zenker M., Hüffmeier U., Thiel C.T., Rüschendorf F., et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. Part A. 2006;140:2063–2074. doi: 10.1002/ajmg.a.31416. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
