

Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders
- PMID: 32878977
- PMCID: PMC7605265
- DOI: 10.1101/gr.266221.120
Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders
Abstract
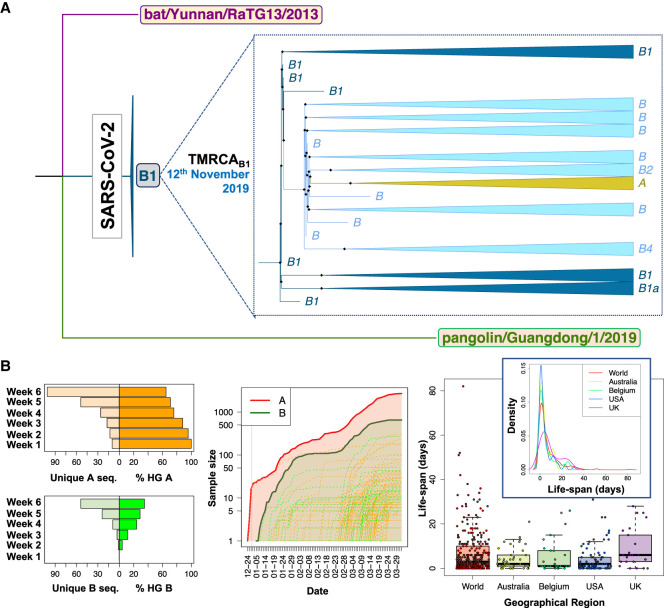
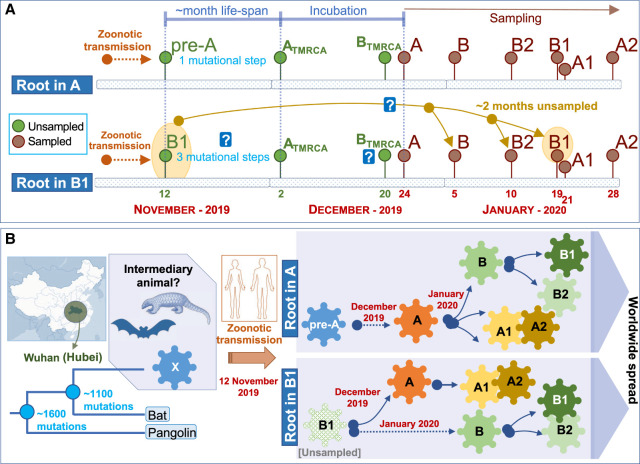
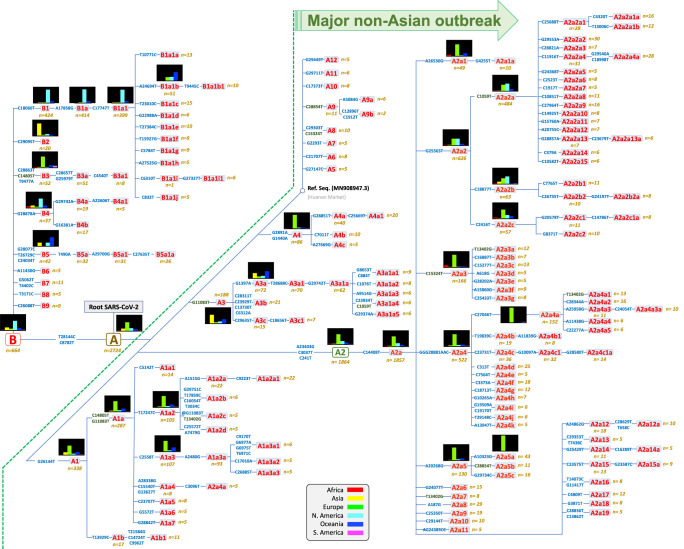
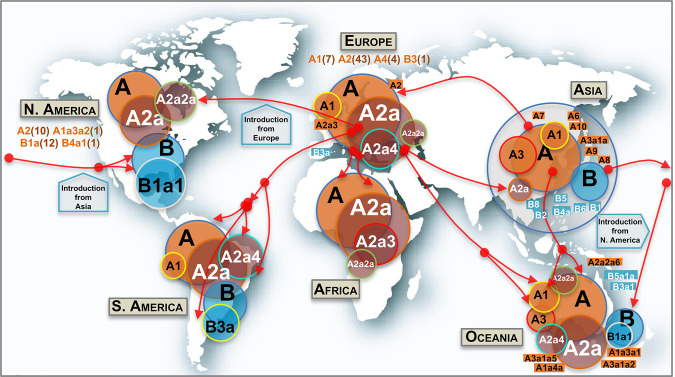
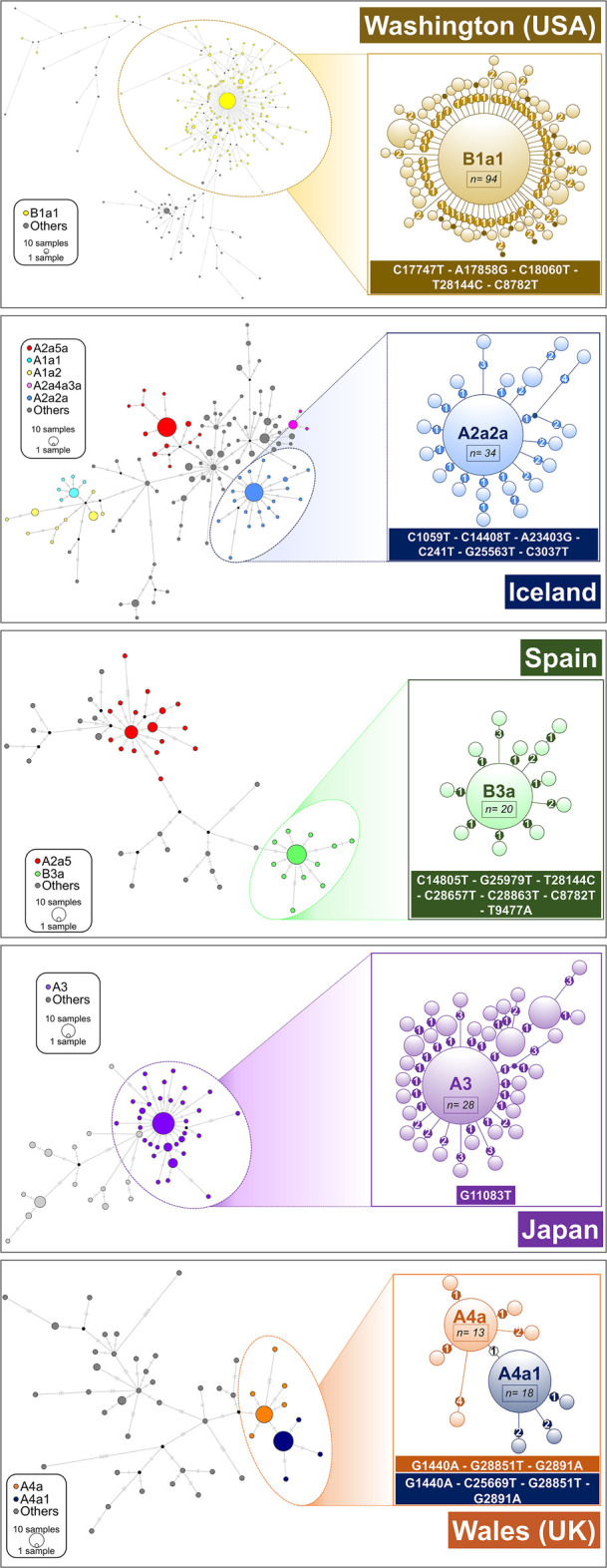
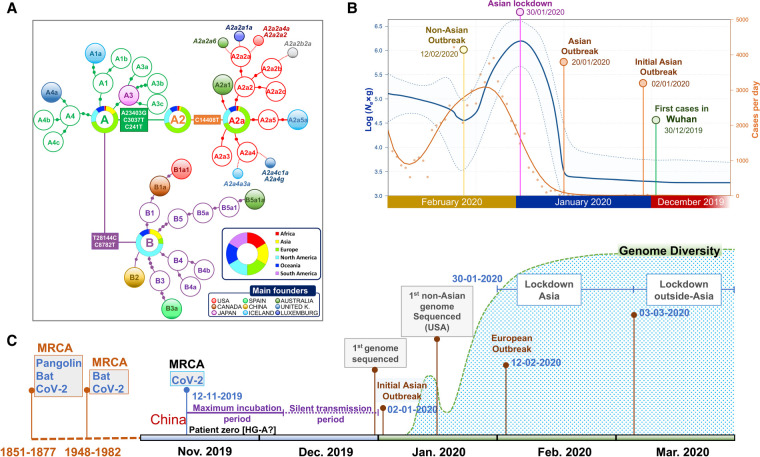
The human pathogen severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the major pandemic of the twenty-first century. We analyzed more than 4700 SARS-CoV-2 genomes and associated metadata retrieved from public repositories. SARS-CoV-2 sequences have a high sequence identity (>99.9%), which drops to >96% when compared to bat coronavirus genome. We built a mutation-annotated reference SARS-CoV-2 phylogeny with two main macro-haplogroups, A and B, both of Asian origin, and more than 160 sub-branches representing virus strains of variable geographical origins worldwide, revealing a rather uniform mutation occurrence along branches that could have implications for diagnostics and the design of future vaccines. Identification of the root of SARS-CoV-2 genomes is not without problems, owing to conflicting interpretations derived from either using the bat coronavirus genomes as an outgroup or relying on the sampling chronology of the SARS-CoV-2 genomes and TMRCA estimates; however, the overall scenario favors haplogroup A as the ancestral node. Phylogenetic analysis indicates a TMRCA for SARS-CoV-2 genomes dating to November 12, 2019, thus matching epidemiological records. Sub-haplogroup A2 most likely originated in Europe from an Asian ancestor and gave rise to subclade A2a, which represents the major non-Asian outbreak, especially in Africa and Europe. Multiple founder effect episodes, most likely associated with super-spreader hosts, might explain COVID-19 pandemic to a large extent.
© 2020 Gómez-Carballa et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Artesi M, Bontems S, Gobbels P, Franckh M, Maes P, Boreux R, Meex C, Melin P, Hayette MP, Bours V, et al. 2020. A recurrent mutation at position 26,340 of SARS-CoV-2 is associated with failure of the E-gene qRT-PCR utilized in a commercial dual-target diagnostic assay. J Clin Microbiol doi.org/10.1128/JCM.01598-20 - DOI - PMC - PubMed
-
- Boni MF, Lemey P, Jiang X, Lam TT, Perry BW, Castoe TA, Rambaut A, Robertson DL. 2020. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol doi.org/10.1038/s41564-020-0771-4 - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous