

Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data
- PMID: 32879317
- PMCID: PMC7468132
- DOI: 10.1038/s41467-020-17967-y
Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data
Abstract
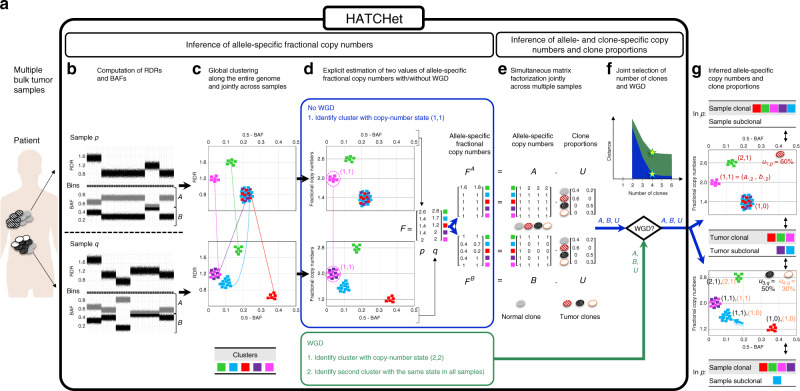
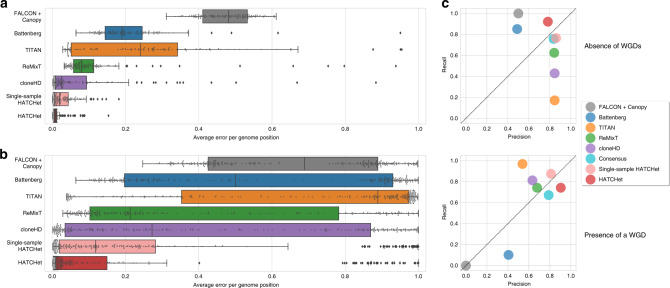
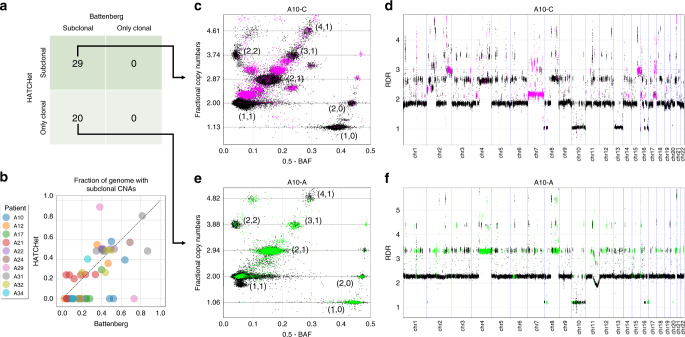
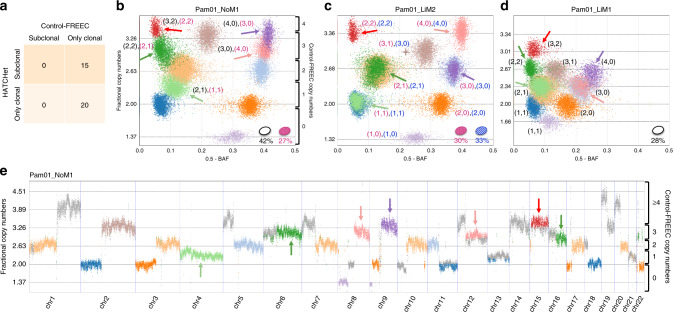
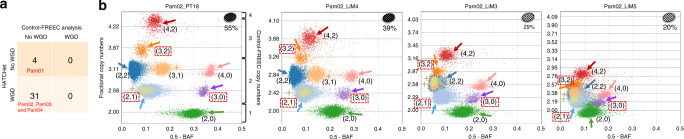
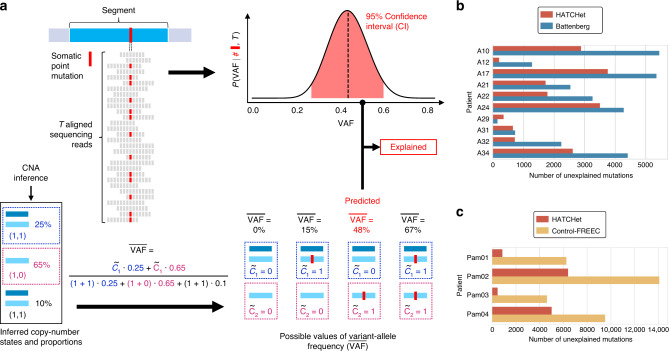
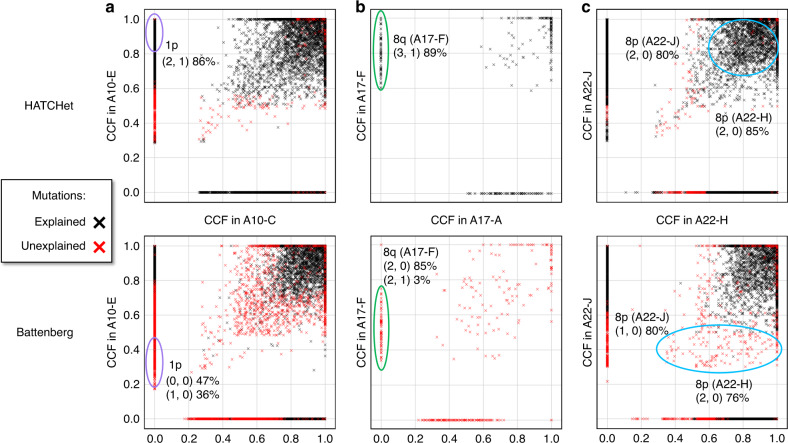
Copy-number aberrations (CNAs) and whole-genome duplications (WGDs) are frequent somatic mutations in cancer but their quantification from DNA sequencing of bulk tumor samples is challenging. Standard methods for CNA inference analyze tumor samples individually; however, DNA sequencing of multiple samples from a cancer patient has recently become more common. We introduce HATCHet (Holistic Allele-specific Tumor Copy-number Heterogeneity), an algorithm that infers allele- and clone-specific CNAs and WGDs jointly across multiple tumor samples from the same patient. We show that HATCHet outperforms current state-of-the-art methods on multi-sample DNA sequencing data that we simulate using MASCoTE (Multiple Allele-specific Simulation of Copy-number Tumor Evolution). Applying HATCHet to 84 tumor samples from 14 prostate and pancreas cancer patients, we identify subclonal CNAs and WGDs that are more plausible than previously published analyses and more consistent with somatic single-nucleotide variants (SNVs) and small indels in the same samples.
Conflict of interest statement
B.J.R. is a cofounder of, and consultant to, Medley Genomics. S.Z. declares no competing interests.
Figures
References
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. - PubMed
-
- Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338. - PubMed
-
- McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
