

Pervasive chromosomal instability and karyotype order in tumour evolution
- PMID: 32879494
- PMCID: PMC7611706
- DOI: 10.1038/s41586-020-2698-6
Pervasive chromosomal instability and karyotype order in tumour evolution
Abstract
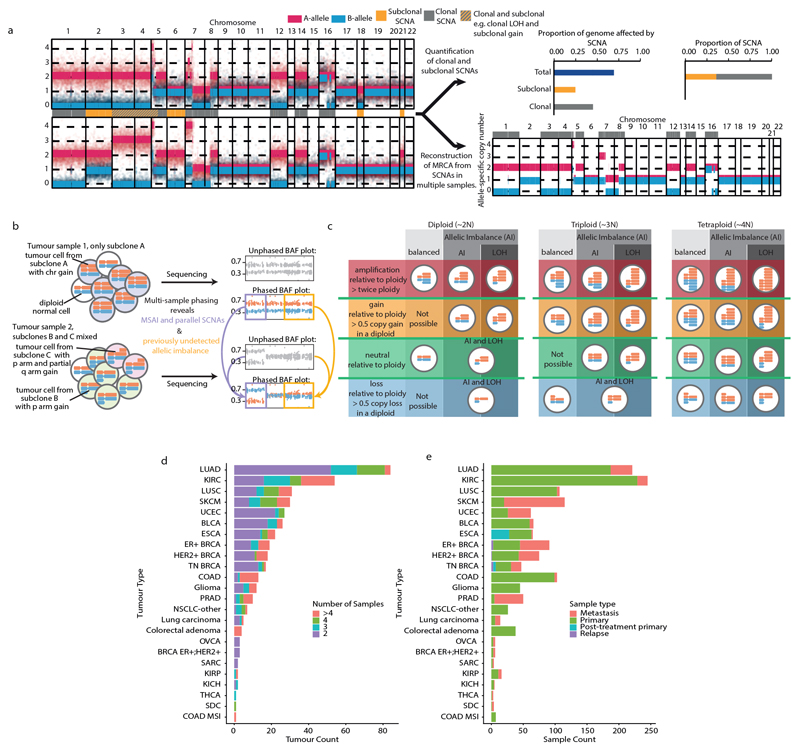
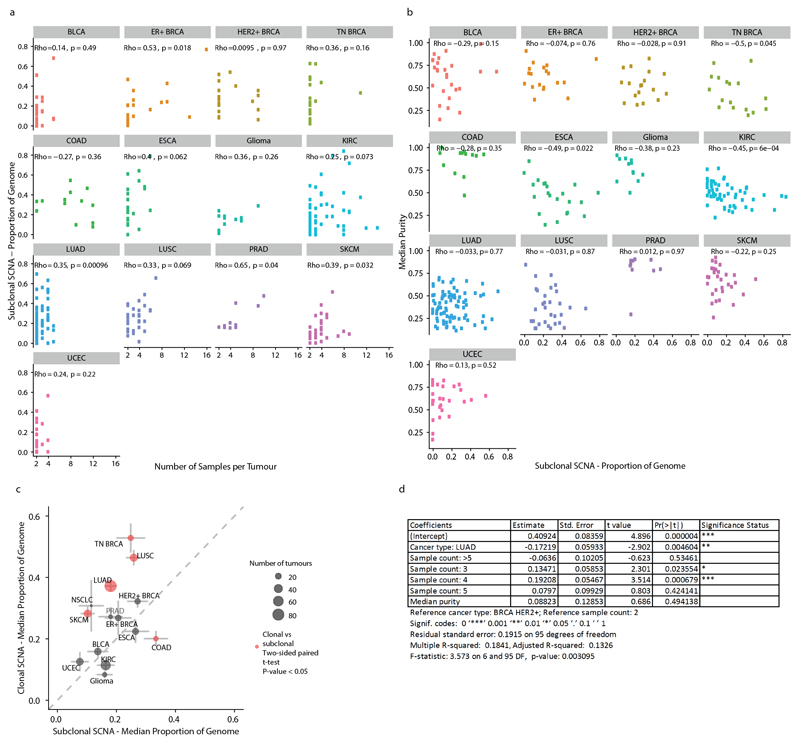
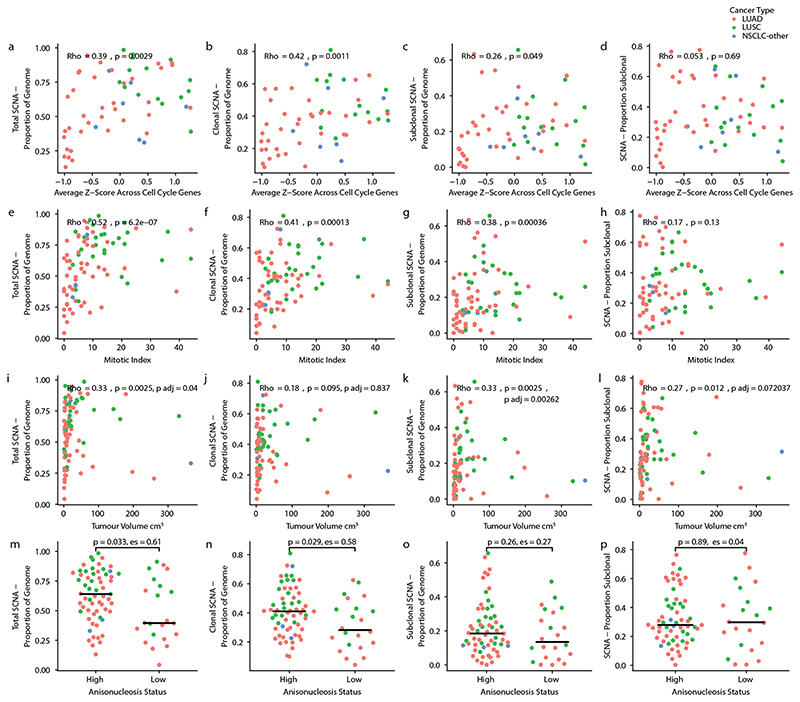
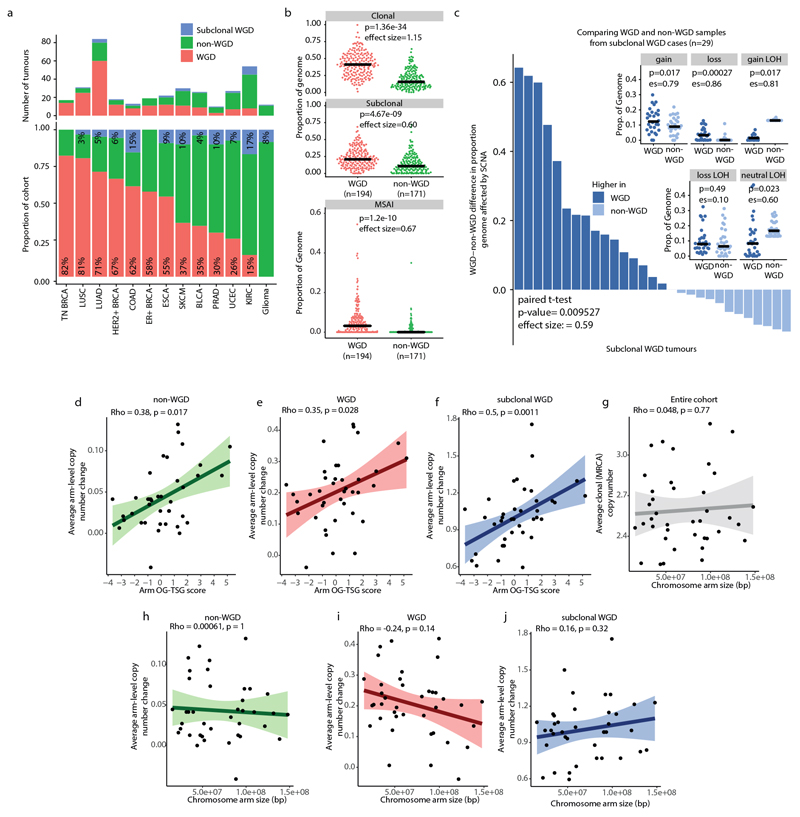
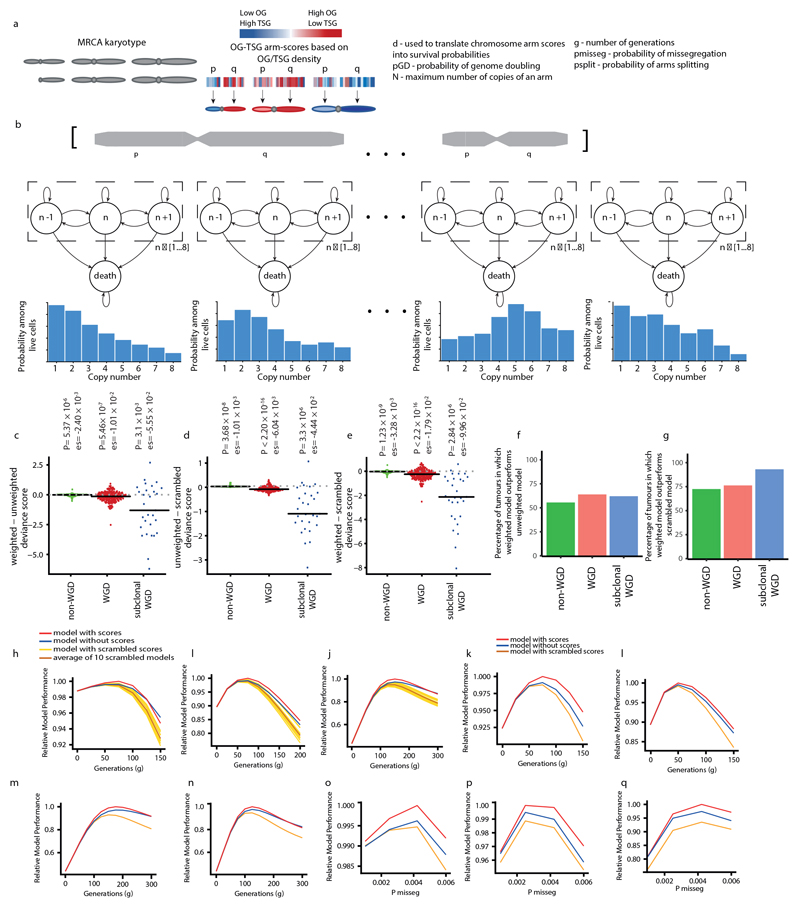
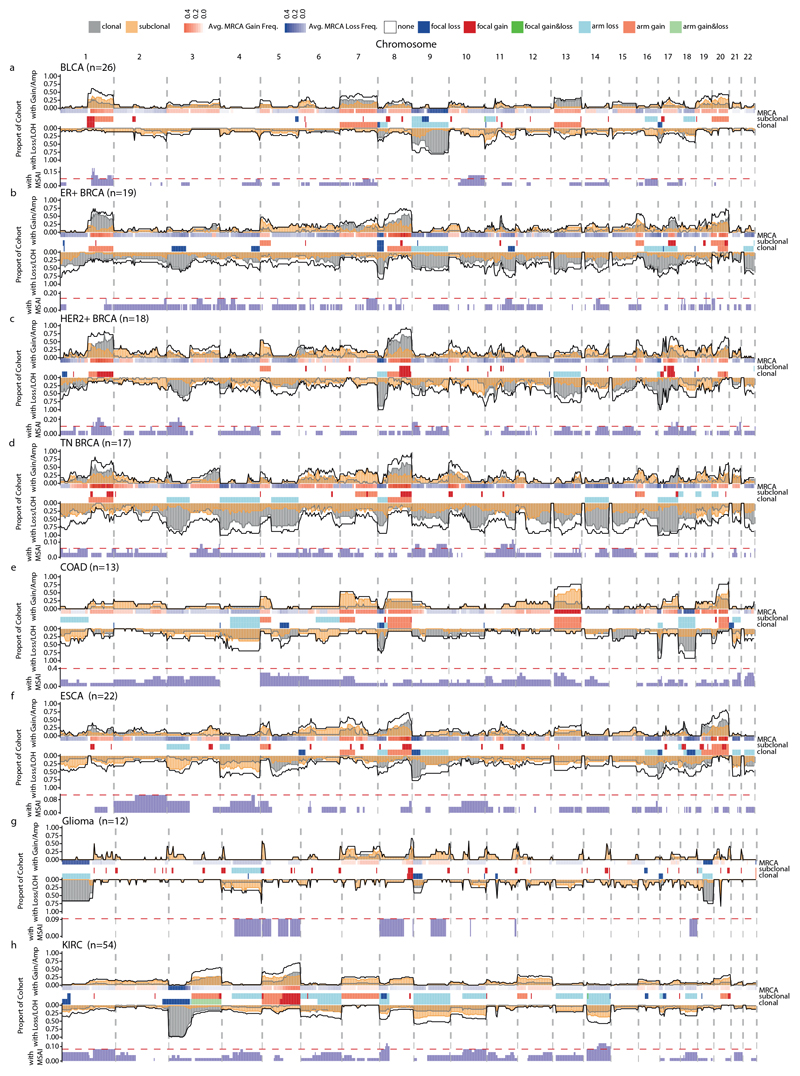
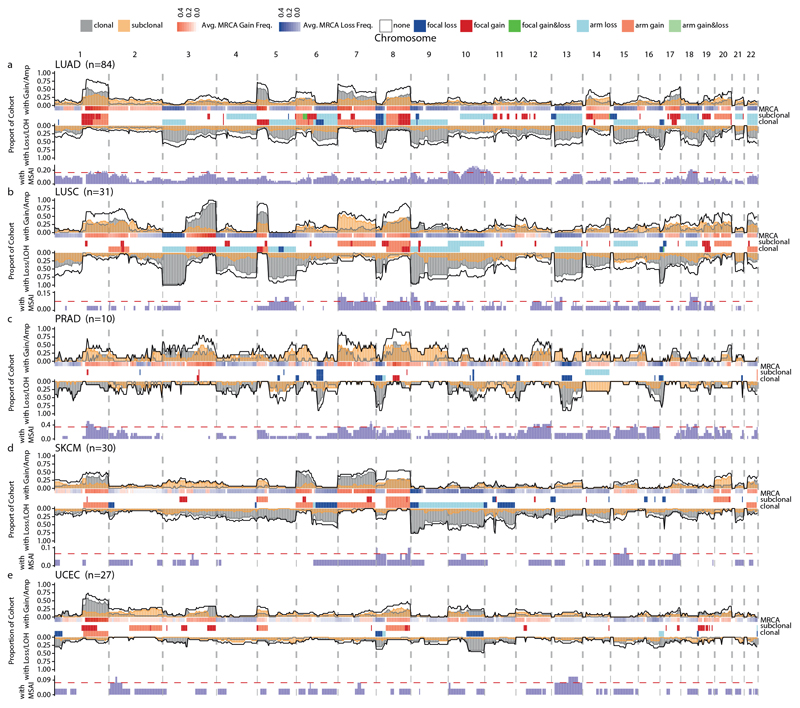
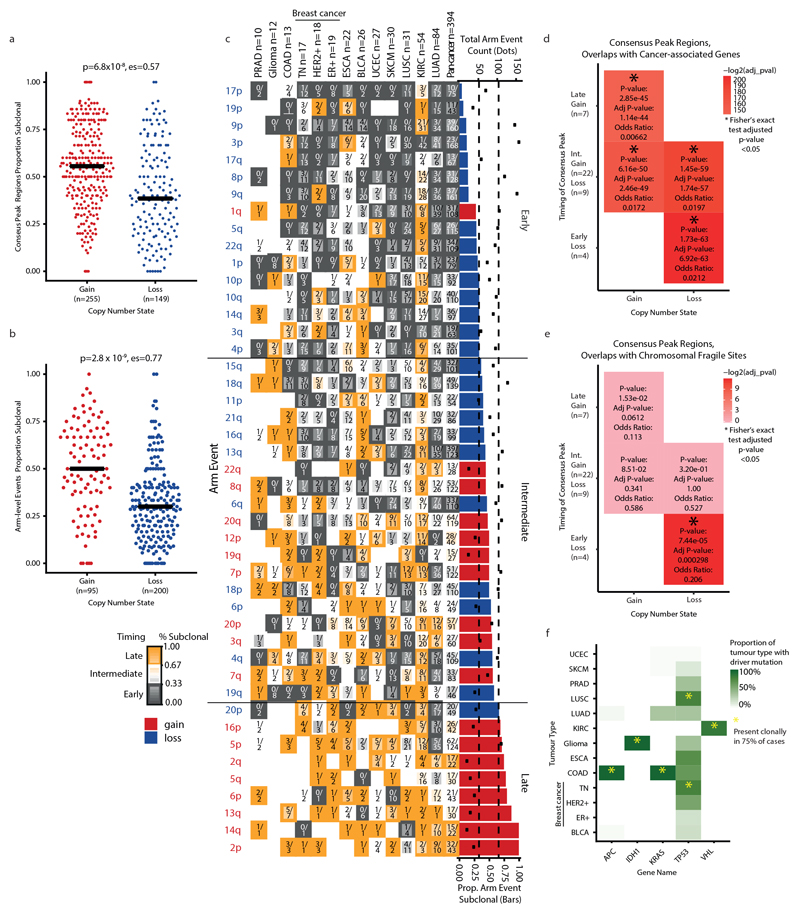
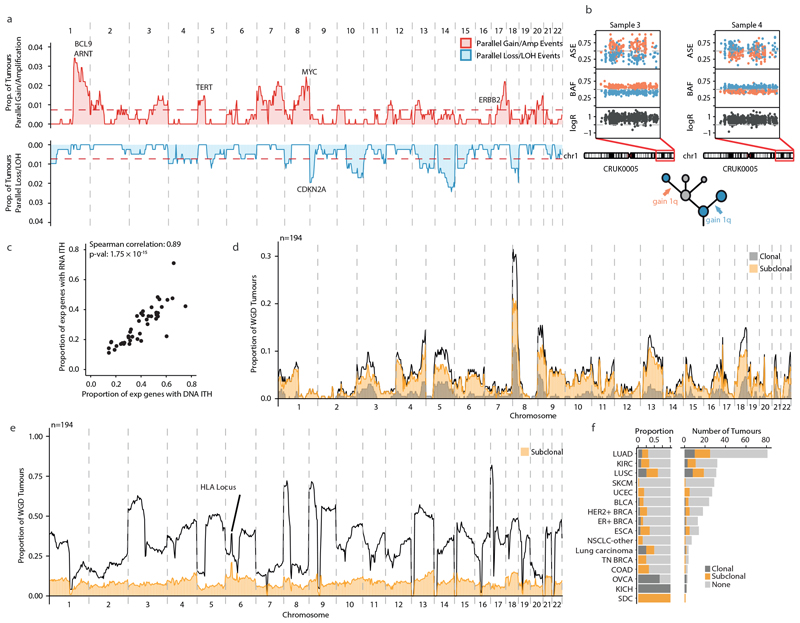
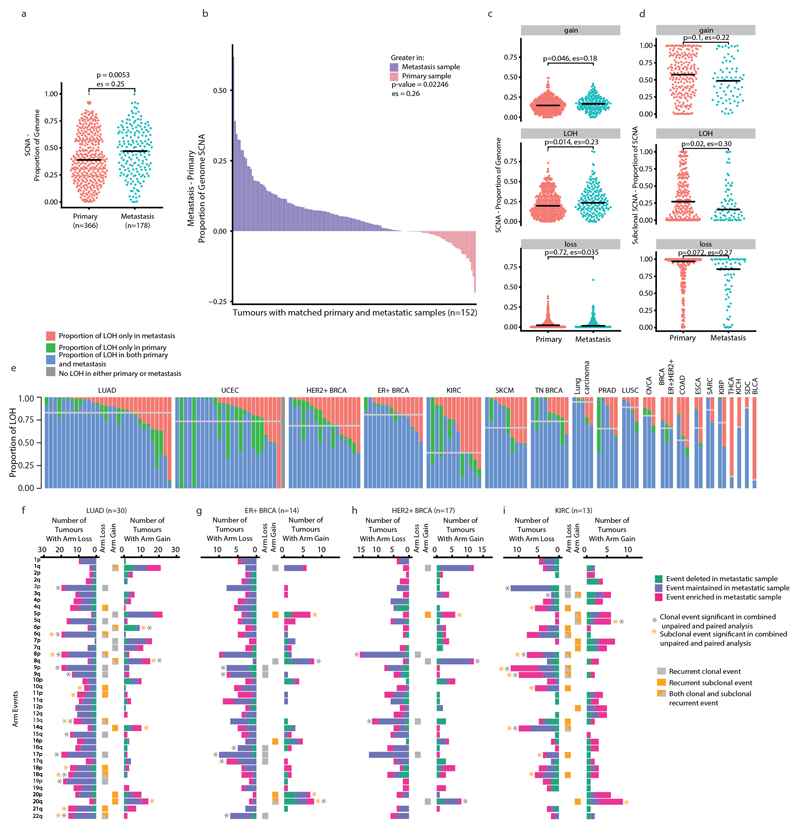
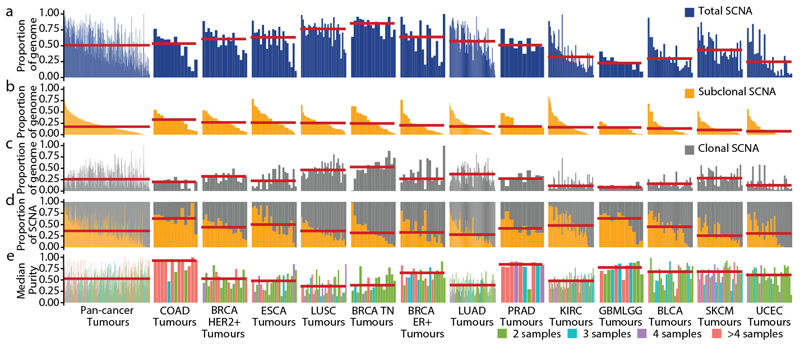
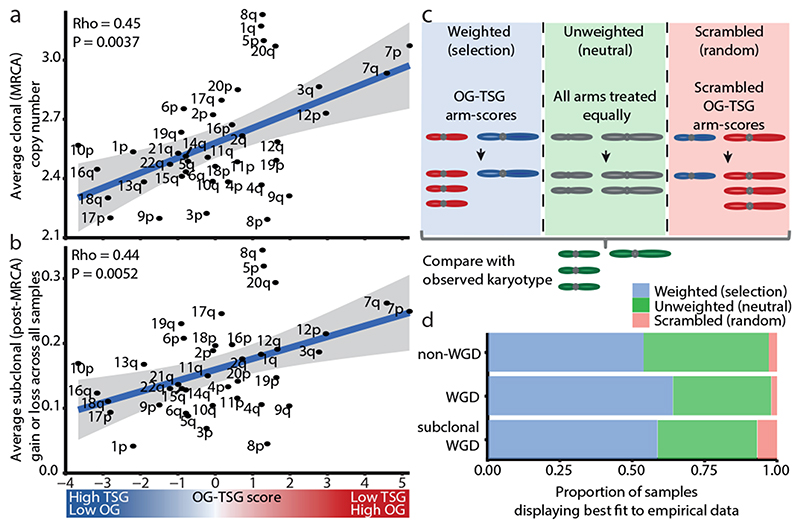
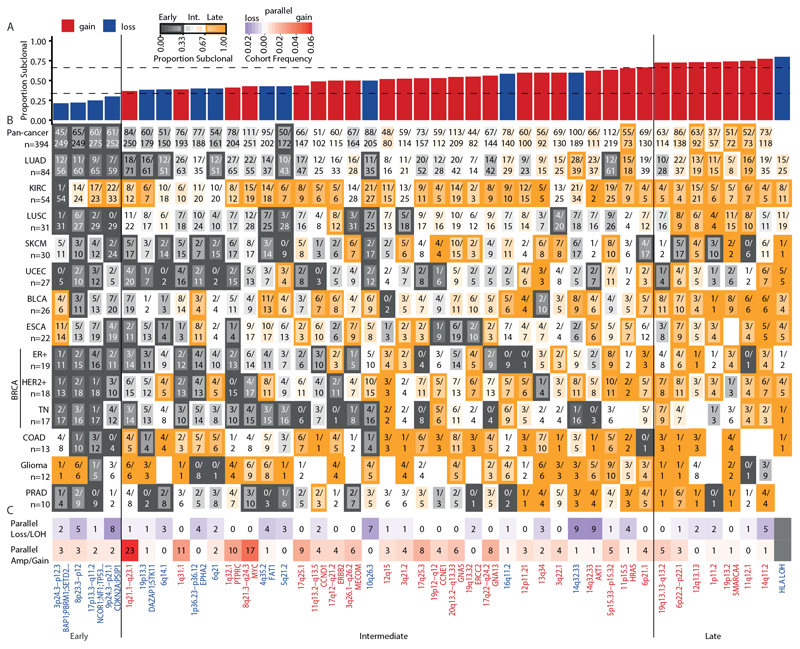
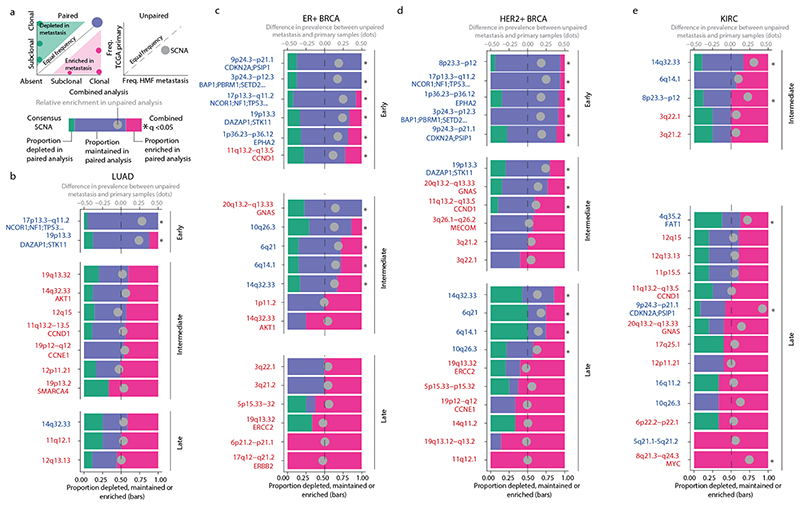
Chromosomal instability in cancer consists of dynamic changes to the number and structure of chromosomes1,2. The resulting diversity in somatic copy number alterations (SCNAs) may provide the variation necessary for tumour evolution1,3,4. Here we use multi-sample phasing and SCNA analysis of 1,421 samples from 394 tumours across 22 tumour types to show that continuous chromosomal instability results in pervasive SCNA heterogeneity. Parallel evolutionary events, which cause disruption in the same genes (such as BCL9, MCL1, ARNT (also known as HIF1B), TERT and MYC) within separate subclones, were present in 37% of tumours. Most recurrent losses probably occurred before whole-genome doubling, that was found as a clonal event in 49% of tumours. However, loss of heterozygosity at the human leukocyte antigen (HLA) locus and loss of chromosome 8p to a single haploid copy recurred at substantial subclonal frequencies, even in tumours with whole-genome doubling, indicating ongoing karyotype remodelling. Focal amplifications that affected chromosomes 1q21 (which encompasses BCL9, MCL1 and ARNT), 5p15.33 (TERT), 11q13.3 (CCND1), 19q12 (CCNE1) and 8q24.1 (MYC) were frequently subclonal yet appeared to be clonal within single samples. Analysis of an independent series of 1,024 metastatic samples revealed that 13 focal SCNAs were enriched in metastatic samples, including gains in chromosome 8q24.1 (encompassing MYC) in clear cell renal cell carcinoma and chromosome 11q13.3 (encompassing CCND1) in HER2+ breast cancer. Chromosomal instability may enable the continuous selection of SCNAs, which are established as ordered events that often occur in parallel, throughout tumour evolution.
Conflict of interest statement
G.A.W. has consulted for and has stock options in Achilles Therapeutics. D.A.M. reports speaker fees from AstraZeneca. M.A.B. has consulted for Achilles Therapeutics. C.V. has received travel expenses from Astellas, Roche and Pfizer, and grant support from Bristol Myers Squibb. R.R. has consulted for and has stock options in Achilles Therapeutics. K.L. reports speaker fees from Roche Tissue Diagnostics. P.K.B. has consulted for Angiochem, Roche-Genentech, Eli Lilly, Tesaro, ElevateBio, Pfizer (Array), and received grant or research support from Merck, Bristol Myers Squibb and Eli Lilly and honoraria from Merck, Roche-Genentech and Eli Lilly. L.D. has sponsored research agreements with C2i-genomics, Natera, AstraZeneca and Ferring, and has an advisory/consulting role at Ferring. P.S. serves an uncompensated consultant for Roche-Genentech. S.L. receives research funding to her institution from Novartis, Bristol Myers Squibb, Merck, Roche-Genentech, Puma Biotechnology, Pfizer, Eli Lilly and Seattle Genetics, has acted as consultant (not compensated) to Seattle Genetics, Pfizer, Novartis, Bristol Myers Squibb, Merck, AstraZeneca and Roche-Genentech and has acted as consultant (paid to her institution) to Aduro Biotech, Novartis, GlaxoSmithKline and G1 Therapeutics. F.A. is a member of the Advisory Boards for Pfizer, AstraZeneca, Eli Lilly, Roche-Genentech, Novartis and Daiichi Sankyo, acknowledges grant support from Pfizer, AstraZeneca, Eli Lilly, Novartis and Daiichi Sankyo and is a co-founder of Pegacsy. V.C.G.T.-H. reports grants and personal fees from Pfizer, Roche, Novartis and Eli Lilly, grants from Eisai and personal fees from Accord. S.T. has received funding from Ventana Medical Systems Inc (grant numbers 10467 and 10530), has received speaking fees from Roche, AstraZeneca, Novartis and Ipsen and has the following European and US patent filed: Indel mutations as a therapeutic target and predictive biomarker (PCTGB2018/051892) and European patent: Clear Cell Renal Cell Carcinoma Biomarkers (P113326GB). M.J.-H. is a member of the Advisory Board for Achilles Therapeutics. S.F.B. holds a patent related to some of the work described targeting CIN and the cGAS-STING pathway in advanced cancer, owns equity in, receives compensation from and serves as a consultant and on the Scientific Advisory Board and Board of Directors of Volastra Therapeutics, and has also consulted for Sanofi, received sponsored travel from the Prostate Cancer Foundation, and both travel and compensation from Cancer Research UK. N.M. has stock options in and has consulted for Achilles Therapeutics and holds a European patent in determining HLA LOH (PCT/GB2018/052004). C.S. acknowledges grant support from Pfizer, AstraZeneca, Bristol Myers Squibb, Roche-Ventana, Boehringer-Ingelheim, Archer Dx Inc (collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceutical, is an AstraZeneca Advisory Board Member and Chief Investigator for the MeRmaiD1 clinical trial, has consulted for Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol Myers Squibb, Celgene, AstraZeneca, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi and the Sarah Cannon Research Institute, has stock options in Apogen Biotechnologies, Epic Bioscience, GRAIL, and has stock options and is co-founder of Achilles Therapeutics. C.S. holds European patents relating to assay technology to detect tumour recurrence (PCT/GB2017/053289); to targeting neoantigens (PCT/EP2016/059401), identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912), a US patent relating to detecting tumour mutations (PCT/US2017/28013) and both a European and US patent related to identifying insertion/deletion mutation targets (PCT/GB2018/051892).
Figures
References
-
- Bolhaqueiro ACF, et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat Genet. 2019;51:824–834. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 211179/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- 21999/CRUK_/Cancer Research UK/United Kingdom
- FC001202/MRC_/Medical Research Council/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- P30 CA008748/CA/NCI NIH HHS/United States
- MR/P014712/1/MRC_/Medical Research Council/United Kingdom
- R01 CA256188/CA/NCI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- FC001202/WT_/Wellcome Trust/United Kingdom
- R35 CA220481/CA/NCI NIH HHS/United States
- ERC_/European Research Council/International
- FC010110/WT_/Wellcome Trust/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- 214584/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- 20466/CRUK_/Cancer Research UK/United Kingdom
- 18176/CRUK_/Cancer Research UK/United Kingdom
- FC001169/MRC_/Medical Research Council/United Kingdom
- FC001169/WT_/Wellcome Trust/United Kingdom
- MR/P014712/2/MRC_/Medical Research Council/United Kingdom
- 17786/CRUK_/Cancer Research UK/United Kingdom
- DP5 OD026395/OD/NIH HHS/United States
- FC001110/CRUK_/Cancer Research UK/United Kingdom
- MR/L016311/1/MRC_/Medical Research Council/United Kingdom
- 29911/CRUK_/Cancer Research UK/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
